
 Español
Español  Português
Português  English
English Carlos J. Galli Mainini
Síndrome de Adams-Stokes. ver Síndrome de Stokes-Adams
Síndrome de Adie. Síndrome neurológico de etiología desconocida. Se caracteriza por una reacción patológica de la pupila en la acomodación (miotonía), por la cual la pupila del lado afectado se contrae y se dilata más lentamente que la del lado sano. Ciertos reflejos tendinosos están ausentes o disminuidos, pero no hay otros trastornos motores o sensitivos, ni signos demostrativos de enfermedad del sistema nervioso central
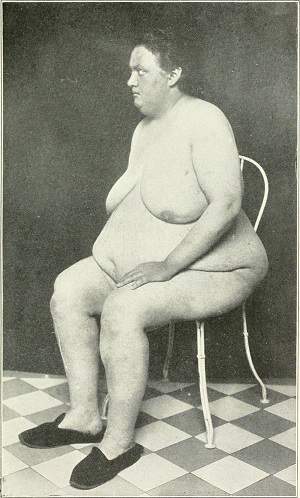
Síndrome adiposogenital de Frohlich. Síndrome endocrinogenital por disfunción hipotalámica. Sus rasgos básicos son: a) obesidad de tipo feminoide con hipoplasia de las gónadas: y ausencia de los caracteres sexuales secundarios; b) diabetes insípida, y c) retardo mental y del crecimiento y trastornos de la visión. Lo producen los tumores u otros procesos patológicos que afectan el área hipotalamohipofisaria y que incluyen el centro hipotalámico del apetito, responsable de la obesidad.
Síndrome adrenogenital. Conjunto de cuadros clínicos producido por hiperfunción suprarrenal y cuyas manifestaciones son predominantemente genitales. Incluye los siguientes: a) el síndrome de hiperplasia suprarrenal congénita. ocasionado por deficiencia específica de enzimas, que intervienen en el metabolismo de los corticosteroides (véase cap.61); b) el síndrome de virilización suprarrenal del adulto, producido por tumores suprarrenales benignos o malignos, y c) el síndrome de feminización suprarrenal, de causa tumoral o por deficiencia enzimática.
Síndrome de la afasia motriz. Síndrome neurológico por oclusión de la división superior de la arteria cerebral media. El cuadro clásico se caracteriza por: a) el individuo conserva su lenguaje interior (sabe lo que tiene que decir) pero ha perdido la memoria de los movimientos necesarios para hacerla, de modo que no puede expresar ese lenguaje en palabras: articuladas (anartria); b) comprende lo que se le dice, y la lectura y la escritura están conservadas; c) es plenamente consciente de lo que le sucede, lo cual a menudo lo lleva a la exasperación, y d) en algunos casos se asocian alteraciones variables de la comprensión. Obedece a lesión de las regiones frontal inferior, parietal anterior e insular anterior, de origen embólico y a veces por tumores o abscesos cerebrales.
Síndrome de la afasia sensorial. Síndrome neurológico por oclusión de la arteria cerebral media izquierda. Resulta de la pérdida de la capacidad de comprender el significado de las palabras (afasia de comprensión), sin alteraciones en la articulación de los vocablos. El individuo queda privado de todo medio de comunicación social. Hay: a) incapacidad del paciente para hacerse entender (ni verbalmente ni por escrito), porque las palabras, correctas del lenguaje son sustituidas por otras de sonido o significado más o menos parecidos (parafasias literal y verbal), o por neologismos o una jerigonza incomprensible; b) incapacidad para encontrar el nombre de los objetos que percibe; c) incapacidad para entender lo que lee o lo que se le dice, y d) sensación, por parte del observador, de que el enfermo no tiene una clara conciencia de lo que le sucede. Obedece a lesiones de la región silviana posterior (temporal y parietal) y habitualmente se produce por embolia.
 Síndrome de Frohlich  Síndrome de la acondroplasia (Suros, Semiología Médica, 1968) |
Síndrome de la agnosia visual. Síndrome neurooftalmológico de origen central. Se produce por lesiones que afectan las vías visuales y las áreas corticales primarias, secundarias, y terciarias de la visión, y el cuadro es el de un individuo que ve bien los objetos, pero que no puede reconocerlos a menos que, según el caso, los toque, los huela o los saboree, o que reconozca los sonidos que emiten.
Síndrome del agujero óptico. Síndrome neurológico de la base del cráneo. Ceguera unilateral, de comienzo insidioso y de carácter progresivo, acompañada o no de dolor homolateral irradiado al cuello y a la región mandibular. Es producido, casi siempre, por un glioma del nervio óptico.
Síndrome del agujero rasgado anterior. ver Síndrome de Bonnet
Síndrome del agujero rasgado posterior. ver Síndrome de Vernet
Síndrome de Ahumada-Del Castillo. ver Síndrome de Forbes-Albright
Síndrome del ala menor del esfenoides. ver Síndrome de Kennedy
Síndrome de Albright-McCune-Sternberg. Síndrome hereditario caracterizado por alteraciones óseas, de la piel y endocrinoginecológicas. Es un cuadro raro que se observa en niñas, y se manifiesta por displasia ósea (engrosamiento, incurvación y fracturas fáciles de los huesos), anomalías pigmentarias de la piel (manchas de bordes irregulares, color café con leche) y pubertad precoz con desarrollo temprano de los caracteres sexuales secundarios.
Síndrome alcohólico fetal. Síndrome infantil debido al abuso de alcohol, por parte de la madre, durante el embarazo. Pueden presentarse los siguientes rasgos, aislados o en combinación: a) talla corta en relación con la que correspondería según el peso; b) malformaciones faciales (hipoplasia del maxilar inferior, paladar hendido) y oculares (microftalmía, pliegue epicanto) y luxación bilateral de cadera; c) anomalías cardíacas y genitales, y d) malformaciones cardíacas y renales. Además, el recién nacido puede desarrollar un cuadro similar; sólo que persistente, al de la abstinencia del alcohol en el adulto, y con el tiempo evidencia un retardo mental de grado variable.
Síndrome de alexia sin agrafia. Síndrome neurológico por lesión del lóbulo occipital. Hemianopsia derecha e imposibilidad de leer en voz alta y de comprender el lenguaje escrito, sin alteraciones en el dictado ni en la lectura espontánea en silencio. Se debe a lesión del lóbulo occipital izquierdo, por lo general tumoral, asociada lesión del cuerpo calloso.
 Síndrome de Albright-McCune-Sternberg Síndrome de Albright-McCune-Sternberg |
Síndrome de Andersen. Síndrome de la mucoviscidosis complicada. Es una tríada de mucoviscidosis (fibrosis quística del páncreas), bronquiectasias e hipovitaminosis A, en la cual el primer componente es la enfermedad de fondo, y los otros dos son complicaciones frecuentes.
Síndrome del ángulo pontocerebeloso. Síndrome neurológico central con compromiso de los nervios craneales V, VII y VIII. Se manifiesta por sordera de percepción (VIII par), seguida de arref1exia o hiporreflexia laberíntica (VIII par), y asociada en ocasiones a arreflexia corneana (V par) y parálisis facial de variable intensidad (VII par). Es causado por meningiomas o neurinomas del nervio acústico.
Síndrome de ansiedad. Síndrome funcional de los estados de ansiedad. Palidez, sudoración, temblor variable, palpitaciones, respiración rápida y superficial o respiración "corta", temor injustificado, etc., sin causa orgánica detectable. Se lo observa en individuos sometidos a situaciones sostenidas de tensión psíquica.
Síndrome por aplastamiento. Síndrome producido por el aplastamiento' sostenido de algún sector del organismo. Define el cuadro de edema, oliguria, hipovolemia, shock, y finalmente, insuficiencia renal aguda, que se observa con frecuencia en individuos que han sufrido lesiones por compresión traumática prolongada, particularmente cuando el traumatismo ha afectado una masa considerable de tejido muscular (p. ej., aplastamiento de ambos miembros inferiores a raíz de un derrumbe).
Síndrome de la apnea del sueño. ver Síndrome de hipersomnia con apnea nocturna
Síndrome de la artería mesentérica superior. Síndrome abdominal ocasionado por obstrucción extrínseca del tránsito duodenal. Lo origina la arteria mesentérica superior al comprimir la tercera porción del duodeno contra el tronco de la aorta abdominal, como consecuencia de lo cual se produce una obstrucción aguda y transitoria, pero recurrente en el tiempo, del segmento en cuestión. Las manifestaciones del síndrome son muy variables: desde perturbaciones mínimas, hasta náuseas y vómitos posprandiales, dolor centroabdominal y, en ocasiones, distensión extrema del estómago y el duodeno.
Síndrome del asa aferente. Síndrome secundario a la gastroyeyunostomía. Se manifiesta por episodios posprandiales recurrentes de náuseas y distensión y dolor en el hemiabdomen superior. El cuadro se atribuye a la obstrucción parcial crónica, de tipo intermitente, del asa intestinal proximal (duodeno y yeyuno)
Síndrome del asa ciega. Síndrome por disfunción del intestino delgado. Se caracteriza, en sus formas severas, por esteatorrea, mal absorción de vitamina B 12 y anemia no ferropénica. Se atribuye a proliferación bacteriana anormal del intestino delgado, como consecuencia de estenosis del órgano o de operaciones que dejan asas intestinales con tránsito poco activo.
Síndrome de Asherrnan. Síndrome de amenorrea por destrucción del endometrio. Amenorrea y esterilidad secundarias, con cicatrices y adherencias intrauterinas, y desaparición más o menos completa del endometrio. Es causado por procedimientos instrumentales (curetaje), habitualmente a raíz de hemorragias posparto o de abortos complicados con infección.
Síndrome de la atetosis doble. ver Síndrome de Vogt.
Síndrome de Aubertin. Síndrome de la interrupción abrupta del flujo sanguíneo en las cavidades cardíacas. Es un cuadro, de aparición brusca y rápidamente progresiva de dolor retroesternal agudo, disnea, taquipnea, angustia, lividez, pulso filiforme, colapso y casi siempre muerte, que se observa como complicación de la estenosis mitral y se produce cuando un coágulo desprendido de la aurícula izquierda ocluye abruptamente el orificio auriculoventricular.
Síndrome auriculotemporal. ver Síndrome de Frey.
Síndrome de Avellis. Síndrome neurológico del tronco cerebral con alteraciones motoras y sensitivas, y compromiso del X nervio craneal. Parálisis de la cuerda vocal y el paladar blando del lado de la lesión (X nervio craneal), y en el lado opuesto hemiplejía que respeta la cara y pérdida de la sensibilidad térmica y dolorosa (lesión del techo del bulbo y del haz piramidal). La causa es tumoral o por reblandecimiento cerebral.
Síndrome de Ayerza. Síndrome de la insuficiencia pulmonar crónica avanzada Cianosis intensa, insuficiencia cardíaca congestiva y grados variables de policitemia.
Síndrome de Babinski-Nageotteo. Síndrome neurológico de la lesión unilateral del bulbo raquídeo. Hemiplejía y hemianestesia del lado opuesto al de la lesión, y hemiasinergia, hemiataxia y lateropulsión del mismo lado de la lesión; y si están afectadas las fibras simpáticas de la formación reticular, también miosis, enoftalmía y ptosis palpebral del lado de la lesión. Obedece en general a una obstrucción vascular por encima del entrecruzamiento del haz piramidal.
Síndrome de Babinski-Vaquez. Síndrome neurológico y vascular de la sífilis avanzada. Es un cuadro variable y polimorfo, que depende de la localización y la gravedad de las lesiones principales y que se manifiesta, básicamente, por alteración de los reflejos tendinosos (arreflexia patelar y aquiliana) y de los de la pupila (arreflexia pupilar, miosis, ete.), anomalías. del líquido cefalorraquídeo, estado demencial e insuficiencia aórtica manifiesta.
Síndrome de Bafuerstedt. Síndrome de la linfadenosis cutánea benigna. Hiperplasia inflamatoria de los folículos linfáticos de la piel, que afecta preferentemente la cara y orejas, de adultos jóvenes, bajo la forma de nódulos solitarios o diseminados y que suele involucionar espontáneamente.
Síndrome de Bandl-Frommel. Síndrome de la rotura inminente del útero. Es propio del período expulsivo del parto y se caracteriza por tetanización del cuerpo uterino y del anillo de Bandl, y por la posibilidad de palpar a través, de la pared abdominal, dos cordones duros y tensos que corresponden a los ligamentos redondos.
Síndrome de Banti. Síndrome de la esplenomegalia crónica congestiva. Obedece a procesos que producen una hipertensión crónica del sistema porta, con hiperesplenismo secundario y se caracteriza por: a) esplenomegalia; b) anemia, leucopenia y trombocitopenia, con hiperplasia de la médula ósea y c) cirrosis hepática, con ascitis y hemorragias por rotura de várices esofágicas.
Síndrome de Bard y Pic. Síndrome del carcinoma de la cabeza del páncreas. Se manifiesta por una tríada característica: ictericia crónica progresiva, gran dilatación de la vesícula biliar y caquexia rápidamente evolutiva. Obedece a compresión del colédoco por la masa tumoral..
 Síndrome de Barraquer-Simmons Síndrome de Barraquer-Simmons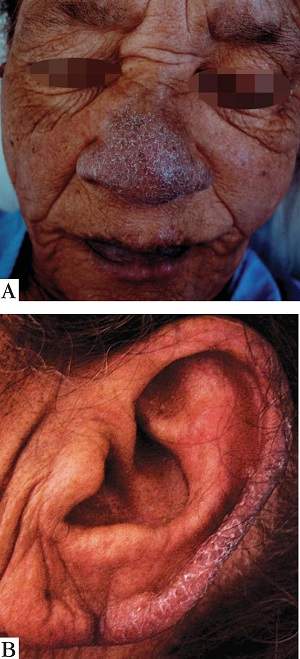 Síndrome de Bazex |
Síndrome de Barraquer-Simmons. Síndrome de la lipodistrofia progresiva. Emaciación profunda de la parte superior del cuerpo, en particular la cara, y gran obesidad en la mitad inferior especialmente en nalgas y muslos. Se la observa en mujeres jóvenes y la causa se desconoce.
Síndrome de Barré-Lieou. Síndrome relacionado con afecciones orgánicas de la columna cervical. Se caracteriza por cefalea, trastornos visuales pasajeros, fenómenos vasomotores en la cara, vértigo, acufenos, parestesias craneofaciales, disfagia y disfonía intermitentes, etc. Es causado por irritación directa de nervios espinales y simpáticos, en pacientes con artrosis avanzada de las tres últimas, vértebras cervicales, o por trastornos vasculares (vertebrobasilares) o psicógenos.
Síndrome de Barrett. Síndrome de la esofagitis crónica. Inflamación crónica de la mucosa del tercio inferior del esófago, asociada o no al desarrollo de úlceras, pépticas, que lleva progresivamente a la estenosis por retracción cicatrizal.. La causa subyacente es, en general, la sustitución del epitelio escamoso o pavimentoso propio del esófago por un epitelio secretor de tipo gástrico.
Síndrome de Bartter. Síndrome de híperaldosteronismo secundario con hiperplasia de las células yuxtaglomerulares. Se caracteriza por alcalosis hipopotasémica y aumento marcado de los niveles plasmáticos de renina, sin hipertensión arterial ni respuesta a la acción presora de la angiotensina. La causa es probablemente genética y el cuadro puede acompañarse de retardo mental y baja estatura.
Síndrome de Bassen-Kornzweig. Síndrome caracterizado por anomalías de las b-lipoproteínas y otras alteraciones. Se manifiesta por ausencia de las -lipoproteínas, plasmáticas, retinitis pigmentaria, acantocitosis de los glóbulos rojos, y trastornos neurológicos progresivos que comienzan en la infancia y se manifiestan por ataxia, arref1exia y alteraciones severas de la sensibilidad propioceptiva. La causa es desconocida.
Síndrome de Bastian. Síndrome de la sección completa de la médula espinal. Incluye una fase inicial aguda, bilateral, con paraplejía y anestesia por debajo del nivel de la lesión, arreflexia cutánea y tendinosa, retención urinaria e incontinencia fecal; y una fase posterior caracterizada por rigidez espástica y recuperación de los reflejos y del tono muscular. La causa es casi siempre traumática.
Síndrome de Bazex. Síndrome de la acroqueratosis paraneoplástica. Consiste en lesiones hiperqueratósicas, con fondo eritematoso, en el pabellón de la oreja, la nariz y los dedos, acompañadas, de espesamiento de las uñas y perionixis. Suele existir una neoplasia de vías aéreas, superiores.
Síndrome de Behcet. Síndrome vasculítico con manifestaciones oculares y mucosas. Cuadro recidivante, de evolución crónica, caracterizado por episodios de conjuntivitis, uveítis y ulceraciones de la córnea y de las mucosas, bucal y genital. El origen es probablemente viral y en las formas, graves conduce a la ceguera.
Síndrome de Benedikt. Síndrome neurológico del tronco cerebral con compromiso del III nervio craneano y manifestaciones motoras y cerebelosas. Es un cuadro de parálisis de la motilidad ocular del lado de la lesión (III nervio craneano), con hemiplejía frustra, hernitemblor, hemiataxia y hemiasinergia del lado opuesto (lesión del núcleo, rojo y de la vía corticoespinal). Lo producen las hemorragias, reblandecimientos y tumores que afectan la región peduncular..
Síndrome de Bernard-Soulier. Síndrome hereditario relacionado con anomalías en el mecanismo de coagulación. Se caracteriza por tendencia a la hemorragia acompañada de trombocitopenia leve, plaquetas gigantes o de morfología atípica, tiempo de coagulación prolongado y consumo de protrombina anormal..
Síndrome de Bemhardt-Roth. Síndrome de la meralgia parestésica. Se manifiesta por parestesias y otros trastornos sensitivos habitualmente leves en la cara lateral de uno de los muslos. Obedece a constricción del nervio femorocutáneo a nivel de su ingreso en la fascia lata.
Síndrome de Bertolotti. Síndrome relacionado con anomalías estructurales de la columna vertebral. Es una tríada constituida por escoliosis, sacralización de la quinta vértebra lumbar y ciática, las dos primeras entendidas como anomalías: básicas y la tercera como complicación secundaria.
Síndrome de Bianchi. Síndrome neurológico por lesión de la corteza parietal. Afasia sensorial, apraxia y alexia, caracterizada por la incapacidad del enfermo de, entender el significado de las palabras que escucha o que lee, como si correspondieran a un idioma desconocido. Es propio de ciertas lesiones, casi siempre tumorales, del lóbulo parietal izquierdo.
Síndrome de Bloch-Sulzberger. Síndrome de alteración del metabolismo pigmentario con anomalías corporales múltiples. Se caracteriza por lesiones cutáneas pigmentadas, vesiculosas y luego verrugosas, asociadas a defectos en el desarrollo de los ojos, huesos y sistema nervioso central. Es hereditario y afecta casi exclusivamente a mujeres.
Síndrome de Blount-Barber. Síndrome relacionado con anomalías estructurales de los miembros. inferiores. Asociación de osteocondrosis de la epífisis superior de la tibia, rotación interna de la tibia sobre el fémur y moderado pie plano. Se debe al desarrollo anómalo, de carácter aislado, de la extremidad proximal de la tibia.
Síndrome de Bogorad. Síndrome que aparece en el período de recuperación de ciertos casos de parálisis facial. El rasgo distintivo es la aparición de lagrimación unilateral, del lado de la parálisis, que se produce cuando el individuo ingiere alimentos. Se atribuye a peculiaridades del proceso de regeneración nerviosa, durante el cual algunas de las fibrillas destinadas a las glándulas salivales se conectarían con otras dirigidas al sistema lagrimal.
Síndrome de Bonnet. Síndrome neurológico de la base del cráneo. Se manifiesta por oftalmoplejía completa, miosis y trastornos motores y sensitivos del nervio trigémino, de comienzo insidioso y avance progresivo. Lo producen los tumores infiltrantes de la base del cráneo.
Síndrome de Bonnier. Síndrome neurológico por lesión de estructuras centrales con compromiso de los nervios craneales III, V, VIII, IX y X. Se manifiesta por estado vertiginoso, debilidad y somnolencia, neuralgia en el territorio del trigémino, alteraciones de la motilidad ocular, inestabilidad e hipoacusia y otras alteraciones. Es producido por lesiones que afectan al núcleo, de Deiters, a las vías vestibulares asociadas y a los pares craneales III, V, VIII, IX y X.
Síndrome de bradiarritmia-taquiarritmia. Síndrome de la alternancia de períodos de bradiarritmia y taquiarritmia. Se caracteriza por la alternancia de períodos de bradiarritmia (30 a 50 latidos/min.) con mareos o síncope, que pueden durar desde minutos a meses, y períodos de taquiarritmia supraventricular (hasta. 200 por minuto), que pueden durar desde minutos a meses y acompañarse de palpitaciones y otros síntomas de una taquicardia supraventricular paroxística. En los intervalos entre unos y otros puede existir un ritmo sinusal normal. La causa es desconocida.
Síndrome de Brissaud-Marie. Síndrome neurológico de la histeria. Hemiespasmo del labio y de la lengua, sin causa que lo justifique, sin sistematización neurológica y en una personalidad con rasgos histéricos.
Síndrome de Brissaud-Sicard. Síndrome 'provocado por lesiones irritativas de la protuberancia. Espasmo de la musculatura facial del lado de la lesión central, con hemiplejía o hemiparesia del lado opuesto al de la lesión.
Síndrome de Bristowe. Síndrome de los tumores del cuerpo calloso. Se caracteriza por hemiplejía progresiva del lado de la lesión, hemiparesia contralateral leve, disfagia, estupor, letargo, coma y muerte.
Síndrome de Brown-Séquard. Síndrome de hemisección de la médula espinal. Parálisis fláccida, atrofia muscular y abolición de la sensibilidad profunda, en el lado de la lesión, con conservación de la sensibilidad superficial, asociada a pérdida de la sensibilidad superficial con conservación de la motricidad y de la sensibilidad profunda en el lado opuesto al de la sección. La causa es casi siempre traumática (proyectiles. de armas de fuego).
Síndrome de Bruns. Síndrome neurológico de los tumores del cuarto ventrículo. Se caracteriza por cefalea intensa e intermitente, vértigo, vómitos y alteraciones de la visión, que se acentúan cuando el paciente realiza un movimiento brusco de la cabeza.
Síndrome de Budd-Chiari. Síndrome de la obstrucción de las venas suprahepáticas. La forma aguda se caracteriza por dolor epigástrico, hepatomegalia dolorosa rápidamente progresiva, ascitis grave e ictericia leve, y conduce a la muerte en un lapso de días o de uno a dos meses. En la forma crónica el comienzo es insidioso y las manifestaciones se acentúan de un modo gradual hepatomegalia con gran esplenomegalia, dolor abdominal, ascitis y edema de los miembros inferiores. La obstrucción se produce por trombosis (endoflebitis, policitemias) o por compresión extrínseca (p-ej., hipernefroma).
Síndrome bulbar. ver Síndrome de Dejerine, def 2.
Síndrome bulbar hernilateral. ver Síndrome de Babinski-Nageotie.
Síndrome bulbar lateral. ver Síndrome de Wallenberg.
Síndrome de Burnett. ver Síndrome de leche y álcalis.
Síndrome de Bumier. Síndrome endocrino y neurológico de origen tumoral. Se caracteriza por enanismo, distrofia adiposogenital y atrofia del nervio óptico y suele ser producido por tumores de crecimiento lento que comprimen la hipófisis y la región del quiasma.
Síndrome de Bywaters. ver Síndrome por aplastamiento.
Síndrome de Camurati-Engelmann. Síndrome de la displasia diafisaria progresiva. Se caracteriza por engrosamiento simétrico y aumento de diámetro de la diáfisis, de los huesos largos, con dolor en las zonas afectadas, fatiga, marcha anormal y atrofia muscular. La causa se desconoce.
Síndrome de Canada-Cronkite. Síndrome de poliposis intestinal con alteraciones orgánicas secundarias. Se caracteriza por la presencia de una poliposis intestinal. múltiple, que da lugar a una enteropatía crónica con esteatorrea. Como consecuencia de ésta, por deficiencias múltiples de nutrientes, se producen alopecia, alteraciones cutáneas y atrofia de las uñas. La causa es desconocida.
Síndrome del canal condíleo. Síndrome neurológico de la base del cráneo. Parálisis unilateral de la lengua producida por la compresión del XII nervio en la base del cráneo por lo general de origen tumoral
Síndrome de Caplan. Síndrome artrítico asociado a la neumoconiosis. Define la asociación de una artritis reumatoidea con la presencia de nódulos pulmonares múltiples (granulomas reumatoideos) en pacientes con neumoconiosis subyacente. El trastorno dificulta el trasporte de gases a través de la pared alveolar.
Síndrome capsulotalámico. Síndrome neurológico por lesión de estructuras subcorticales. Se caracteriza por hemianopsia, hemianestesia, hemiplejía parcial, alteraciones en la percepción del dolor e inestabilidad emocional. Es causado por lesiones vasculares o tumores del tálamo óptico y de la cápsula interna.
Síndrome del carcinoide maligno. Síndrome de los tumores carcinoide del intestino delgado. Se caracteriza, típicamente, por episodios paroxísticos de enrojecimiento cianótico de la cara y el cuello, cólicos abdominales, diarrea hipotensión y espasmo bronquial; y en fases más avanzadas por alteraciones pigmentarias, ascitis, lesiones del endocardio e insuficiencia cardíaca. Suelen existir metástasis hepáticas múltiples y el cuadro obedece a la hipersecreción de serotonina por las células tumorales.
Síndrome centromedular. Síndrome neurológico por lesión de la médula cervical. Se caracteriza por paresia variable de los cuatro miembros, pero desproporcionadamente mayor en los miembros superiores. Es causado por lesiones de la porción central de la médula cervical, de origen vascular o degenerativo.
Síndrome centroposterior de la médula espinal. Síndrome neurológico por lesión de la médula espinal. Se manifiesta por trastornos vasomotores y por disociación de la sensibilidad de tipo siringomiélico (pérdida de la sensibilidad térmica y dolorosa, con conservación de la sensibilidad táctil y profunda). Lo producen las lesiones, de la porción central posterior de la sustancia gris de la médula espinal
Síndrome cerebral postraumático. Síndrome residual alejado de los traumatismos craneanos. El cuadro es polimorfo predominantemente subjetivo, pero de presentación frecuente en este tipo de pacientes y se manifiesta por cefalea de intensidad y localización variables, amnesia, vértigo, tinnitus; palpitaciones, fatigabilidad, irritabilidad, insomnio y dificultad de concentración.
Síndrome cervical. Síndrome doloroso relacionado con alteraciones orgánicas de la columna cervical. Se caracteriza por dolor del cuello de presentación periódica, que se irradia a la espalda, el hombro y/o el miembro superior. Puede ser simétrico o unilateral, y suele exacerbarse con los movimientos de rotación o extensión de la cabeza. Obedece a compresión o irritación de las raíces de los nervios cervicales por pinzamientos artrósicos.
Síndrome de Cestan. Síndrome neurológico por lesión extensa de estructuras cerebrales. Hemiplejía y hemianestesia del lado opuesto al de la lesión, con hemiasinergia, laringoplejía, lateropulsión y síndrome de Horner del mismo lado de la lesión. El cuadro obedece a lesiones dispersas que afectan la pirámide, las vías sensitivas el pedúnculo cerebeloso inferior, el núcleo ambiguo y el centro oculopupilar.
Síndrome de la cimitarra. Síndrome radiológico originado por la desembocadura anómala de las venas pulmonares. Se lo observa en las radiografías frontales del tórax y consiste en una modificación de la imagen cardíaca, que se hace convexa a la derecha (por desembocadura de las venas pulmonares derechas en la vena cava inferior) y cóncava a la izquierda (por desplazamiento del corazón hacia el pulmón derecho que es hipotrófico). Globalmente la imagen recuerda a la de una cimitarra, en la cual, la hoja está representada por el corazón y el mango por los grandes vasos.
Síndrome de Claude Bernard-Horner. Síndrome de la parálisis del simpático cervical. Se caracteriza por hundimiento del globo ocular (enoftalmía), ptosis del párpado superior con ligera elevación del párpado inferior y estrechamiento de la hendidura palpebral, miosis, anhidrosis, y congestión vascular del lado afectado de la cara. La causa suele ser tumoral (por compresión).
Síndrome de Claude-Lhermitte. Síndrome neurológico por compresión del infundíbulo y del tercer ventrículo. Se manifiesta por un cuadro complejo, polimorfo, que incluye narcolepsia o hipersomnia, alteraciones vasomotoras y de la regulación térmica, diabetes insípida y una diversidad de síntomas: adicionales. La causa es generalmente tumoral.
Síndrome del clic sistólico-soplo telesistólico. Síndrome del prolapso de la válvula mitral. Se caracteriza por la auscultación de un clic sistólico tardío, en la meso o telesístole, seguido de un soplo sistólico de tonalidad alta, en crescendo-decrescendo, por regurgitación a través de una de las valvas de la mitral. El paciente puede tener síntomas (episodios de taquicardia, palpitaciones, dolor precordial indefinido, etc.) o no. Se trata de un trastorno habitualmente benigno que predomina en mujeres, jóvenes.
Síndrome de Clouston. Síndrome de la displasia ectodérmica hidrótica. Se caracteriza por hiperqueratosis palmoplantar, hiperpigmentación cutánea, hipotricosis, destrucción de las uñas, y a menudo retardo mental. La trasmisión es autosómica dominante.
Síndrome de coagulación íntravascular diseminada. Síndrome asociado a trombosis y hemorragias diseminadas a nivel de la microcirculación. Es un cuadro de microtrombosis generalizada acompañado de hemorragias en napa, a menudo irreversibles, que obedece a la intervención de múltiples mecanismos superpuestos: consumo y agotamiento de factores de la coagulación, daño generalizado del endotelio capilar, fibrinólisis exagerada, estado de shock, etc. El proceso se acompaña de trombocitopenia, fibrinogenopenia y exceso de productos de degradación de la fibrina en el torrente circulatorio. Se lo observa en quemados y accidentados graves, en el shock séptico, en la leucemia, y en otras condiciones de similar gravedad.
Síndrome de Cogan., Síndrome vertiginoso asociado a trastornos de la visión y la audición. Presencia de una queratitis intersticial no sifilítica reversible, acompañada de vértigo, zumbidos, nistagmo y sordera rápidamente evolutiva, irreversible. Afecta a adultos jóvenes y la causa es desconocida.
Síndrome de la cola de caballo. Síndrome neurológico por compresión de las raíces lumbares y sacras. Paraplejía fláccida, arreflexia tendinosa, anestesia dolorosa en silla de montar (anal, perineal, genital y glútea), trastornos esfinterianos e impotencia. Se produce por traumatismos o por compresión tumoral.
Síndrome del cólera pancreático. ver Síndrome WDHA.
Síndrome del colon irritable. Síndrome de una afección crónica benigna del intestino grueso. Se caracteriza por alteraciones de la motilidad intestinal, con cólicos y períodos de estreñimiento y diarrea, y habitualmente secreción exagerada de moco, Se lo considera un trastorno funcional.
Síndrome. de Collet-Sicard. Síndrome neurológico de la base del cráneo. El cuadro es idéntico al del síndrome de Villaret (lesión unilateral de los nervios craneanos IX, X, XI y XII), pero sin parálisis del simpático cervical. Las: causas también son las mismas.,
Síndrome por compresión de la arteria vertebral. ver Síndrome del vértigo cervical
Síndrome del conducto auditivo interno. Síndrome neurosensorial de la lesión del nervio auditivo. Sordera de percepción, hipoestesia corneana e hiporreflexia o arreflexia vestibular. La causa más frecuente es el neurinoma del nervio auditivo.
Síndrome del conducto cístico remanente. ver Síndrome del muñon cístico
Síndrome del conducto lumbar estrecho. Síndrome neurológico periférico por irritación de raíces nerviosas. Se caracteriza por lumbalgia acompañada de radiculalgia crural o ciática, que aparece o se agrava mucho al caminar (claudicación intermitente radicular); en ocasiones hay parestesias de pies, paresia muscular y discretos trastornos esfinterianos. Es causado por estrechez congénita del conducto lumbar, pero suele manifestarse en general después de los 50 años, debido al agregado de otros procesos patológicos, (osteofitosis, etc.).
Síndrome del cono medular. Síndrome neurológico de las lesiones del segmento terminal de la médula. Anestesia pudenda que en ocasiones tiene una distribución en "silla de montar'; incontinencia urinaria y fecal por disfunción de los esfínteres; abolición de la eyaculación y la erección; y pérdida de los reflejos anal y plantar. La causa suele ser tumoral o traumática.
Síndrome del corazón pulmonar agudo. Síndrome de la insuficiencia circulatoria aguda de origen pulmonar. Sobreviene bruscamente y se manifiesta por disnea intensa, cianosis o lividez, hipertensión venosa con ingurgitación yugular e hipotensión arterial sistémica, dilatación de la arteria pulmonar, y desviación del eje eléctrico del ECG a la derecha. La mortalidad es elevada y se produce por embolia o trombosis de la arteria pulmonar o de sus ramas.
Síndrome del corazón pulmonar crónico. Síndrome de insuficiencia cardíaca derecha por hipertensión prolongada de la circulación menor en las neuropatías crónicas. Se caracteriza por disnea de esfuerzo, con o sin accesos asmatíformes, tos crónica, hipocratismo digital, cianosis cefalea y somnolencia; en las fases avanzadas hepatomegalia congestiva y edemas periféricos. Hay poliglobulia, signos radiológicos de la enfermedad pulmonar subyacente y cambios electrocardiográficos típicos (onda P alta y picuda en II, III y aVF y negativa en aVL, onda R alta en VI, etc.). Se lo observa en el enfisema, la bronquitis crónica y las bronquiectasias, la tuberculosis pulmonar y otras, afecciones.
Síndrome del cordón anterior. Síndrome neurológico por lesión medular. La lesión se localiza en la porción anterior de la médula espinal, de modo que hay hipoalgesia, hipoestesia y parálisis completa por debajo de este nivel, con preservación relativa de las funciones que dependen del cordón posterior (sensibilidad táctil, vibratoria y de posición). La causa suele ser traumática (proyectiles de arma de fuego).
Síndrome coreico. Síndrome neurológico por lesión de los núcleos grises de la base. Movimientos musculares anormales, amplios, anárquicos, involuntarios e irreprimibles, como los de una extraña danza, que no obedecen a sistematización alguna y que desaparecen durante las horas de sueño; se asocia a hipotonía muscular. Existe una forma crónica hereditaria progresiva (carea de Huntington) y otra aguda que acompaña al embarazo o a ciertos casos de fiebre reumática (carea de Sydenham). Es causado por lesión de los núcleos grises de la base (caudado, putamen, pálido, centro de Luys y locus niger).
Síndrome coronario intermedio., Denominación general de los cuadros de isquemia coronaria que no corresponden a un infarto de miocardio, ni se ajustan al patrón característico de la angina de pecho. Incluye tres síntomas diferentes: a) el síndrome de la insuficiencia coronaria aguda, que se caracteriza por la aparición aislada de un episodio doloroso intenso, a menudo en reposo, acompañado de alteraciones electrocardiográficas: típicas: en la onda T y en el segmento ST, pero sin modificaciones en el complejo QRS; b) el síndrome de la angina inestable similar al anterior pero definido por la presentación de episodios repetidos durante el reposo, y c) el síndrome de la angina progresiva o preinfarto, en el cual los episodios aparecen en respuesta a esfuerzos cada vez menos intensos; o con más; frecuencia, o con una duración más prolongada, o con modificación cualitativa de sus características.
Síndrome costal del enfisema pulmonar. Síndrome doloroso asociada, a enfermedad pulmonar obstructiva crónica. Dolor constante que el paciente refiere al perímetro de la base del tórax, y que suele intensificarse durante las complicaciones habituales en esta afección (bronquitis agudas; etc.). Se atribuye a factores estructurales (hiperexpansión de la caja torácica, osteoporosis senil, etc.) y al esfuerzo muscular permanente que el individuo realiza para respirar.
Síndrome de Costen. Síndrome de la disfunción temporomasticatoria. Se caracteriza por dolor de localización temporomaxilar, acompañado de crujidos de la articulación correspondiente, lateralización del maxilar inferior al abrir la boca, y en ocasiones trismo; también puede haber síntomas auditivos (dolor, hipoacusia, zumbidos). Obedece a maloc1usión de la articulación temporomaxilar, con irritación de los nervios de la cuerda del tímpano y auriculotemporal.
Síndrome de la costilla cervical. ver Síndrome del estrecho superior del tórax.
Síndrome costoclavicular. ver Síndrome del estrecho superior del tórax.
Síndrome de Courvoisier-Terrier. Síndrome del carcinoma de la ampolla de Váter. El cuadro se caracteriza por ictericia, acolia y coluria, asociadas a una marcada dilatación de la vesícula biliar, pero sin la rápida caquexia que acompaña al carcinoma de la cabeza del páncreas,
Síndrome CREST. Síndrome asociado a la esclerodermia. Su nombre deriva de las manifestaciones principales del cuadro: C de calcicosis, debido a la presencia de depósitos cálcicos dispersos a nivel subcutáneo y periarticular; R de Raynaud, por la frecuencia con que el fenómeno de Raynaud aparece en las fases iniciales de la enfermedad; ES de esclerodactilia, por el predominio en los dedos de la rigidez que afecta a todo, el organismo, y T de telangiectasia, por el desarrollo de este tipo de anomalías en la superficie cutánea. Se lo observa, en general, en las formas menos severas de la esclerosis sistémica progresiva.
 Síndrome de Crigler-Najjar Síndrome de Crigler-Najjar |
Síndrome de Crigler-Najjar. Síndrome neurológico relacionado con anomalías congénitas en el metabolismo de la bilirrubina. Se caracteriza por las manifestaciones propias de la impregnación ictérica de los núcleos de la base (kernicterus) (severos trastornos neurológicos), acompañadas de niveles elevados de bilirrubina no conjugada en la sangre. La enfermedad se evidencia desde el nacimiento, suele llevar a la muerte durante el primer año de vida, y la causa es una deficiencia hereditaria de glucuroniltrasferasa,
Síndrome de Cruveilhier-Baumgarten. Síndrome de la hipoplasia congénita del sistema de las venas suprahepáticas. El cuadro es causado por la oclusión de las ramas intrahepáticas de las venas señaladas, y sus manifestaciones, que son graves, derivan del daño orgánico directo que produce la malformación (hepatopatía fibrosa crónica); de la repercusión hemodinámica resultante (hipertensión portal, con várices esofágicas, notable esplenomegalia e hiperesplenismo); de la función compensadora que ejercen las venas umbilicales, que no se colapsan con el nacimiento (circulación colateral hacia la vena cava superior, soplo suave, continuo, auscultable en la región periumbilical), y de las eventuales complicaciones (insuficiencia hepática, hemorragias digestivas, etc.). A diferencia de otros procesos de características similares, no se acompaña de ascitis.
Síndrome del cuerpo calloso ver Síndrome de Bristowe.
Síndrome cutáneo paraneoplásico. Síndrome cutáneo asociado a procesos tumorales de otras regiones del organismo. Incluye una variedad de cuadros que preceden o acompañan a la aparición de las manifestaciones clínicas de un tumor, entre ellos la acantosis nigricans (cáncer gástrico, y con menos frecuencia, tumores intestinales, pulmonares o ginecológicos), dermatomiositis (neoplasias mamarias o genitales, y en ocasiones digestivas o respiratorias), ictiosis adquirida (enfermedad de Hodgkin y afecciones hematopoyéticas malignas), acroqueratosis de Bazex (cáncer de laringe), etcétera.
 Síndrome de Chotzen |
Síndrome de Cyriax. Síndrome doloroso de los cartílagos costales. Se caracteriza por dolor de intensidad variable en la región paraesternal que con relativa frecuencia se irradia al cuello, el hombro y/o el brazo. El cuadro es similar al de la angina de pecho, del cual se diferencia por su carácter persistente y porque se exacerba al palpar uno o más de los cartílagos costales, en donde radica la causa del problema.
Síndrome de Charlin. Síndrome de la neuralgia del nervio nasal. Sus rasgos son: 1) dolores intensos, unilaterales, en crisis paroxísticas, en una región nasoorbitaria, 2) hidrorrea homolateral, y 3) lesiones tróficas corneales. Obedece a una neuritis causada por afecciones de la nariz.
Síndrome de Chauffard-Still ver Síndrome de Still.
Síndrome de Chiari-Frommel. Variedad del síndrome de la galactorrea no puerperal. Es propio del posparto muy alejado y se caracteriza por lactación prolongada, amenorrea y atrofia genital, en el primer, caso por hipersecreción sostenida de prolactina, y en los dos restantes por la disminución concomitante en la secreción de gonadotrofinas pituitarias y como consecuencia de esto, de estrógenos por parte del ovario. La causa no se conoce con precisión, pero se la ubica en la hipófisis misma o en el hipotálamo.
Síndrome de Chotzen. Síndrome hereditario caracterizado por malformaciones predominantemente craneales y digitales. Designa la combinación de acrocefalia con polidactilia y sindactilia parcial, a las que a veces se agregan malformaciones faciales (hipertelorismo ocular) y retardo mental. La afección se trasmite como un rasgo autosómico dominante.
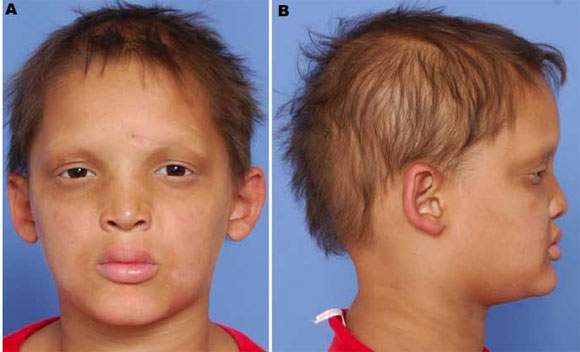
Síndrome de Christ-Siemens. Síndrome de la displasia ectodérmica anhidrótica. Se caracteriza por: a) piel lisa y lustrosa, ausencia de glándulas sudoríparas y deficiente formación de pelos; b) nariz en silla de montar, y frente y mentón prominentes; c) ausencia de gusto y olfato; d) anomalías dentarias y e) retardo mental. Se trasmite como rasgo ligado a X.
 Síndrome de Churg-Strauss |
Síndrome de Churg-Strauss. Síndrome de la granulomatosis alérgica. Eosinofilia prominente, infiltrados pulmonares difusos y, asociación con asma bronquial. Es una vasculitis similar. a la de la poliarteritis nodosa, que compromete, además, al corazón, riñón, intestino y nervios periféricos.
Síndrome de Da Costa. Síndrome de la astenia neurocirculatoria. Complejo sintomático crónico 'caracterizado por sofocación, vahidos, sensación de fatiga, dolor precordial y palpitaciones, sin alteraciones cardíacas orgánicas. Es de naturaleza emocional y se lo llama también corazón de soldado.
Síndrome de Dandy-Walker. Síndrome neurológico resultante de un obstáculo congénito. en la circulación' del líquido cefalorraquídeo. Se manifiesta desde el nacimiento por hipertensión endocraneana, hidrocefalia y trastornos predominantemente cerebelosos. La causa es una atresia congénita de los agujeros de Magendie y de Luschka del cuarto ventrículo, que impide el drenaje normal del líquido cefalorraquídeo.
Síndrome de Debré-Sémélaigne. Síndrome del cretinismo hipotiroideo con alteraciones musculares. Asociación de cretinismo hereditario aumento del volumen muscular, y lentitud de la contracción de los músculos y de los reflejos tendinosos. Es de causa genética y se trasmite como un rasgo autosómico recesivo.
Síndrome de deficiencia de ácido nicotínico. Síndrome de la carencia de un nutriente esencial. En su forma típica (pelagra) se caracteriza por manifestaciones digestivas (diarrea náuseas y vómitos), cutáneas: (pigmentación facial, eritema de la base del cuello) y mucosas (atrofia de las papilas; linguales, lengua escarlata, rugosa y fisurada), y en los casos graves, por alteraciones neurológicas. El trastorno es raro, pero puede observárselo en pacientes con cáncer o con diarrea crónica.
Síndrome de deficiencia de adenosina desaminasa. Síndrome de inmunodeficiencia congénita con anomalías osteocartilaginosas severas. Se manifiesta en los primeros 6 meses de vida por: a) vómitos, diarrea y detención del desarrollo; b) anomalías metafisarias, deformación en copa de las uniones costocondrales y extremidades cortas; y c) infecciones recurrentes severas; predominantemente respiratorias, cutáneas y digestivas, ocasionadas por virus, bacterias, hongos o protozoarios. En la sangre circulante se observa linfopenia, con acentuada disminución de los linfocitos T, ya veces también de los linfocitos B.
Síndrome de deficiencia aislada de inmunoglobulina M. Síndrome de inmunodeficiencia de carácter hereditario. Se manifiesta por niveles bajos de IgM circulante, con trastornos atópicos y gastrointestinales, esplenomegalia y tendencia al desarrollo de infecciones recurrentes severas; y de procesos malignos. El trastorno es familiar y cuatro veces más frecuente en el hombre que en la mujer.
Síndrome de deficiencia de aldolasa hepática. Síndrome producido por una anomalía congénita en el metabolismo de la fructosa. Se caracteriza por detención precoz del desarrollo, tendencia marcada a la hipoglucemia, eliminación de fructosa con la orina, trastornos renales, y alteraciones hepáticas que en los casos graves terminan en una cirrosis con ascitis y esplenomegalia. La causa es una deficiencia congénita de aldolasa en el hígado.
 Síndrome de Christ-Siemens |
Síndrome de deficiencia de fosforilasa-b-quinasa. Síndrome hereditario con alteraciones hepáticas y manifestaciones hipoglucémicas. Se caracteriza por hepatomegalia, hipoglucemia de ayuno y retardo de crecimiento. Es un cuadro benigno que se trasmite como un rasgo ligado a X y cuyas manifestaciones suelen desaparecer en la adolescencia.
Síndrome de deficiencia de hierro. Síndrome de la carencia de un nutriente esencial. Incluye manifestaciones generales (astenia, fatigabilidad, hipersensibilidad al frío, etc.) alteraciones mucocutáneas; (atrofia de las papilas linguales, estomatitis angular, sequedad de la piel, etc.), y fundamentalmente palidez y anemia con microcitosis e hipocromía, disminución del hierro sérico, aumento de la capacidad de combinación del hierro, y ausencia de este mineral en los extendidos de médula ósea. Es causado por carencias absolutas o relativas (dieta láctea, embarazos múltiples), por pérdida exagerada (hemorragia digestiva oculta, hipermenorrea) o por una combinación de estos mecanismos.
Síndrome de deficiencia de purina nucleósido fosforilasa. Síndrome de inmunodeficiencia congénita con anomalías hematológicas y del tejido conjuntivo. El cuadro no se manifiesta al nacer sino después del primer año de vida, fundamentalmente por infecciones recurrentes asociadas a una disminución progresiva de los linfocitos T de la sangre circulante, y a veces con alteraciones de los tejidos de sostén y anemia hemolítica. Es de causa hereditaria y se trasmite como un rasgo autosómico recesivo.
Síndrome de deficiencia de vitamina A. Síndrome de la carencia de un nutriente esencial. Se caracteriza por alteraciones cutáneas (queratosis, xerodermia, hiperqueratosis folicular), trastornos oculares (xeroftalmía, astenopía, dificultad para la visión nocturna) y facilidad para contraer infecciones, particularmente de las vías respiratorias. El cuadro se observa rara vez en su forma pura, debido a la abundancia de la vitamina A en los alimentos, pero sí como componente de los estados severos de desnutrición.
Síndrome de deficiencia de vitamina B. (tiamina). Síndrome de la carencia de un nutriente específico. Da lugar al beriberi e incluye manifestaciones generales (astenia, anorexia, etc.), trastornos neuromusculares (debilidad de los miembros inferiores; polineuritis; con arreflexia o hiporreflexia patelar y parestesias, etc.) y alteraciones cardiovasculares en la variante húmeda de la afección (cardiomegalia, insuficiencia cardíaca congestiva, etc.). Se lo observa como componente de cuadros multicarenciales, con cierta frecuencia en la desnutrición del alcohólico, y ocasionalmente en lactantes y en el embarazo.
Síndrome de deficiencia de vitamina B2 (riboflavina). Síndrome de la carencia de un nutriente esencial. Se manifiesta por alteraciones cutáneas (queilitis, estomatitis anular; disfunción sebácea, etc.) y de las mucosas (glositis, atrofia de las papilas linguales), y por trastornos oculares (astenopía, ambliopía, vascularización de la córnea). Se lo observa, casi siempre, 'como componente de cuadros multicarenciales.
Síndrome de deficiencia de vitamina B6 (piridoxina). Síndrome de la carencia de un nutriente esencial. Incluye alteraciones mucocutáneas. (glositis seborrea nasolabial), trastornos neurológicos (neuropatía periférica, que predomina en los miembros inferiores), y en ciertos casos de la infancia, accesos convulsivos resistentes a la medicación habitual. Se lo observa casi siempre como componente de cuadros multicarenciales.
Síndrome de deficiencia de vitamina B12 (cianocobalamina). Síndrome de la carencia de un nutriente esencial. Da lugar al cuadro de la anemia perniciosa, que incluye alteraciones cutáneas y mucosas (palidez, ictericia leve de color amarillo limón, glositis), manifestaciones neurológicas (parestesias, ataxia, arreflexia tendinosa, nemitis óptica) y anomalías hematológicas (anemia hipercrómica, megalocitosis, presencia de megaloblastos en médula ósea). Es causada por un aporte insuficiente de la vitamina o por carencia de factor intrínseco gástrico (que es indispensable para su absorción), sea como consecuencia de una gastrectomía o de origen constitucional (con acolia e hipoclorhidria).
Síndrome de deficiencia de vitamina C (ácido ascórbico). Síndrome de la carencia de un nutriente esencial. La forma clásica es el escorbuto y se manifiesta por fatigabilidad y perturbaciones emocionales, tumefacción y sangrado gingival, y dolorimiento muscular en el caso del adulto; y en el niño por dolor y tumefacción de los miembros inferiores, alteraciones radiológicas de los huesos (hemorragias. subperiósticas, aspecto de vidrio esmerilado, etc.), tumefacción y sangrado gingival, y hemorragias en otras regiones del organismo. Es una deficiencia actualmente muy rara, que puede afectar a individuos que viven solos y en ocasiones a lactantes.
Síndrome de deficiencia de vitamina D (colecalciferol). Síndrome de la carencia de un nutriente esencial. Se manifiesta, en el niño, por el complejo sintomático del raquitismo (craneotabes, dentición retardada, deformaciones pelvianas y de la caja torácica, incurvación anormal de las piernas, etc.); y en el adulto por tetania y por osteomalacia que predomina en el cráneo, pelvis; y rodillas. El cuadro se observa en casos de carencia alimentaria (con deficiente producción endógena) de pérdida exagerada (síndromes de mal absorción) o en los raquitismos resistentes a la vitamina D (insuficiencia renal crónica, alteraciones tubulares renales, etc.).
Síndrome de deficiencia de vitamina K (menadiona). Síndrome de la carencia de un nutriente esencial. En el recién nacido se manifiesta por hematomas craneanos o del esternocleidomastoideo, hemorragias meníngeas, bucales o del cordón umbilical, y lesiones purpúricas; aparece a los dos o tres días de nacer y se debe a la imposibilidad de absorber la vitamina K por ausencia de la flora intestinal normal. En el adulto se evidencia por petequias, equimosis, hematomas o hemorragias, y por lo general es consecuencia del uso terapéutico de anticoagulantes cumarínicos.
Síndrome de Dejean. Síndrome neurooftálmico por afecciones del piso de la órbita. Incluye exoftalmía y diplopía por desplazamiento del globo ocular; dolor intenso en la región del maxilar superior, y entumecimiento en el área inervada por las dos primeras ramas del trigémino (oftálmica y maxilar superior). La causa suele ser tumoral.
Síndrome de Déjerine. Síndrome neurológico de las lesiones de la corteza sensorial. Se manifiesta por la pérdida de una variedad de funciones altamente especializadas, de modo que el paciente es incapaz de reconocer por el tacto un objeto conocido (astereognosia), de reconocer cambios en la posición de segmentos corporales o diferencias en la intensidad de los estímulos, etc. Es causado por lesiones vasculares (embolia) y en ocasiones por tumores. // 2. Síndrome neurológico asociado a lesiones focales de la porción anterior del bulbo. En el lado opuesto al de la lesión existe hemiplejía que respeta la cara, mientras que del lado de la lesión se observa parálisis y atrofia de la lengua (XIT nervio) cuando la lesión es superior, o parálisis del velo del paladar y de la cuerda vocal (X nervio) cuando se ubica en un nivel.
Síndrome de Déjerine-Klumpke. Síndrome de la parálisis braquial de tipo inferior. Es una parálisis atrófica de los músculos de la mano y los dedos, asociada a un síndrome de Claude. Bernard-Homer. Obedece a lesión de las raíces C8 y DI, habitualmente por compresión.
Síndrome de Déjerine-Sottas. Síndrome de la polineuropatía hipertrófica progresiva. Se manifiesta inicia1mente por dolor y parestesias en los pies, y luego por debilidad y atrofia de los segmentos distales de los miembros, arreflexia tendinosa e invalidez temprana. Los nervios están algo engrosados por el depósito exagerado, de material colágeno. El trastorno es hereditario y se trasmite como un rasgo autosómico recesivo.
Síndrome de Del Castillo-Trabucco-de la Balze. Síndrome asociado a la ausencia congénita del epitelio germinativo. Se caracteriza por esterilidad y testículos pequeños, con normalidad de la libido y de los caracteres sexuales secundarios. El testículo contiene células de Leydig y de Sertoli normales, pero no células germinales.
Síndrome de Dercum. Síndrome de la adiposis dolorosa. Presencia de depósitos grasos circunscritos, dolorosos, en el tejido subcutáneo de las extremidades y a veces de otros sectores del cuerpo. Suele ser de aparición esporádica, con algunos casos documentados de incidencia familiar.
Síndrome de desacondicionamiento cardiovascular. Síndrome ergométrico de los individuos normales no entrenados. Se manifiesta durante las pruebas de esfuerzo controladas por frecuencia cardíaca y presión arterial que se elevan normal y muy rápidamente, sin alteraciones electrocardiográficas significativas y con una capacidad de esfuerzo mediocre. Representa la condición ideal para indicar ejercicios físicos
Síndrome de descerebración. Síndrome neurológico por autonomía del tronco cerebral. Se caracteriza por hiperextensión de los miembros inferiores con marcada espasticidad, flexión fija de los brazos sobre la pared del tórax, hiperextensión de la cabeza divergencia ocular, y estado de coma en el cual solo se mantienen las funciones vegetativas. Obedece a lesiones que interrumpen la conducción nerviosa a nivel de los tubérculos cuadrigéminos, de origen traumático, tumoral o por hemorragias del tercer ventrículo.
Síndrome de De Toni-Debré-Fanconi. ver Síndrome de Fanconi.
Síndrome de la diarrea acuosa. ver Síndrome WDHA.
Síndrome de Di Ferrante. Síndrome de la mucopolisacaridosis VIII Forma parecida a la de los síndromes de Morquio o de Sanfilippo, con retardo estatural y mental, hepatomegalia y esplenomegalia discretas, hipoacusia y gránulos metacromáticos en los leucocitos.
Síndrome de Dighton-Adair. Síndrome de la osteogénesis imperfecta. Sus rasgos básicos son: a) osteoporosis generalizada con marcada fragilidad de los huesos, y con el tiempo desarrollo de deformaciones óseas múltiples producidas por osteomalacia y por la cicatrización de las fracturas; b) escleróticas azules, y c) sordera por osteoesclerosis, en alrededor de la tercera parte de los casos. Los niveles de calcio y fósforo en sangre y orina son normales. El cuadro es familiar y se trasmite como un rasgo autosómico dominante.
Síndrome de la dilatación aguda del estómago. Síndrome de los posoperatorios complicadas. Vómitos abundantes y continuos, distensión abdominal rápida y progresiva chapoteo, y presencia de un gran nivel líquido en la radiografía simple del abdomen, efectuada en posición de pie. Se produce en individuos que han sido sometidos a una intervención quirúrgica intraabdominal, cuando no se les ha realizado aspiración gástrica mediante una sonda.
Síndrome de disartria-mano torpe. Síndrome neurológico lacunar por lesión de la rodilla y el brazo anterior de la cápsula interna. Se caracteriza por disartria, parálisis facial y paresia de la lengua, acompañados de ligera debilidad y torpeza de los movimientos de la mano. La causa es vascular (trombosis, embolia) en el territorio lenticuloestriado.
Síndrome del disco intervertebral. Síndrome neuromuscular por compresión radicular. Se caracteriza por dolor habitualmente intenso en la región lumbar, que aparece durante o poco después de un esfuerzo intenso en flexión o hiperextensión del tronco sobre los miembros inferiores, y que característicamente se exacerba con los movimientos y maniobras: que aumentan la presión del líquido cefalorraquídeo (tos, defecación, etc.). El dolor irradia a menudo al territorio del ciático (cara posterior del muslo y porciones laterales de la pierna y el pie) y en los casos severos se acompaña de hiporreflexia o arref1exia aquiliana y de signos de degeneración nerviosa en el electromiograma. Obedece a la compresión de una raíz nerviosa por protrusión de un disco intervertebral lumbar.
Síndrome de disfagia y disfonía. Síndrome neurológico por lesión del neumogástrico. Se caracteriza por alteraciones unilaterales que incluyen parálisis del paladar blando, pérdida del reflejo nauseoso y parálisis de la cuerda vocal. Obedece a una lesión intracraneana, por lo general de origen tumoral.
Síndrome de disfunción placentaria. Síndrome del sufrimiento fetal intrauterino. Se caracteriza por pigmentación amarilla intensa del unto sebáceo, la piel y las uñas del feto, pigmentación verdosa del cordón umbilical, asociadas a gigantismo y con frecuencia a muerte del feto antes de nacer. Es propio de los embarazos que se prolongan más allá de los trescientos días, y se produce por degeneración de las estructuras placentarias.
Síndrome de disfunción temporomasticatoria. ver Síndrome de Costen.
Síndrome de disgenesia gonadal. ver Síndrome de Turner
Síndrome de la disqueratosis congénita. Síndrome hereditario caracterizado por alteraciones cutáneas y tendencia al desarrollo de procesos malignos. Se define por la presencia de hiperpigmentación reticu1ar, hiperqueratosis palmar y plantar, leucoplasia de las mucosas y pérdida de las uñas; además hay pancitopenia, y una elevada tendencia al desarrollo de carcinomas. Se trasmite como un carácter ligado al sexo.
Síndrome de la distrofia muscular posmenopáusica. Síndrome miopático de etiología desconocida Se caracteriza por debilidad progresiva de la musculatura proximal de los miembros inferiores y superiores, con dificultad creciente para realizar movimientos y actividades habituales.
Síndrome de la distrofia simpática refleja. Síndrome doloroso acompañado de alteraciones musculares, tróficas y vasomotoras. Aparece en una extremidad poco tiempo después de una situación patológica desencadenante (infarto de miocardio, traumatismo, afección neurológica) y se manifiesta por tumefacción distal (p. ej. de toda una mano o todo un pie), dolor intenso y quemante en la misma región, y labilidad vasomotora con episodios de vasoconstricción o vasodilatación. Puede mantenerse sin cambios durante días o semanas y luego revertir, por completo, o bien evolucionar progresivamente hasta producir alteraciones tróficas severas, atrofia muscular y contractura en flexión. La causa no ha sido establecida con precisión, aunque en general se le atribuye un origen simpático reflejo.
Síndrome de Down. Síndrome genético caracterizado por retardo mental y anomalías típicas. Sus rasgos básicos son: a) estatura baja y cráneo pequeño particularmente en su diámetro anteroposterior; b) facies característica con nariz pequeña, ojos oblicuos y lengua grande y saliente; c) dedo meñique corto, separación excesiva entre los primeros dos dedos de las manos y los pies, y presencia de pliegues palmares simiescos; d) presencia de anomalías esqueléticas leves, y con bastan te frecuencia, de malformaciones cardiovasculares, y e) retardo mental de leve a severo. Obedece a una trisomía del cromo soma 21, de aparición espontánea y más raramente familiar.
Síndrome de Dresbach. Síndrome de la eliptocitosis hereditaria. Se caracteriza por la presencia de eritrocitos ovales o elípticos, con hemólisis poco acentuada, esplenomegalia leve y escasas manifestaciones clínicas. Se trasmite en forma autosómica dominante y obedece a un defecto congénito en el citoesqueleto eritrocitario.
Síndrome de Dressler. Síndrome febril doloroso de ciertos pacientes con infarto de miocardio. Se manifiesta por un cuadro febril que aparece de 1 a 6 semanas después de un infarto de miocardio y se acompaña de dolor retroesternal con las características del dolor pleuropericárdico: irradia al cuello y a ambos hombros, alivia cuando el individuo inclina el torso hacia adelante, y se exacerba con la inspiración profunda. La causa es una pleuropericarditis, a menudo asociada a una neumonitis, y se lo considera como una reacción auto inmune.
Síndrome de Duane. Síndrome congénito caracterizado por alteraciones oculares unilaterales. Se manifiesta por imposibilidad o dificultad acentuada para la rotación externa del ojo, dificultad para la rotación interna, retracción del globo ocular y convergencia defectuosa. El trastorno, es hereditario y se trasmite como un rasgo autosómico dominante.
Síndrome de Duchenne. Síndrome neurológico de la parálisis bulbar progresiva. Es un cuadro progresivo de disartria, disfonía y parálisis, precedida de espasmo de los músculos de los labios y la lengua, debido al creciente compromiso de los nervios craneales V, VII; IX, X y Xll. La afección es de tipo degenerativo y su causa se desconoce.
Síndrome del dumping. ver Síndrome del vaciamiento rápido.
Síndrome de Duplay. Síndrome de la tendinitis calcificante. Consiste en dolor y limitación de los movimientos de la articulación del hombro, por inflamación y calcificación de la bolsa subacromial o subdeltoidea.
Síndrome de Eaton-Lambert. Síndrome miasténico asociado a ciertas neoplasias pulmonares. Se manifiesta por debilidad de las porciones proximales de los miembros superiores e inferiores, acompañada de dolor, entumecimiento y arreflexia o hiporreflexia tendinosa. A diferencia del síndrome de la miastenia grave, a) no hay alteraciones de la musculatura ocular, y b) la fuerza tiende a aumentar, y no a disminuir, con la repetición de movimientos musculares. Por lo general, el cuadro precede en meses, o hasta en dos años, a la aparición de un carcinoma pulmonar. de células, pequeñas.
Síndrome eclámptico. Síndrome edematoso hipertensivo del embarazo. Se manifiesta por la tríada de hipertensión, edema y albuminuria, además de náuseas y vómitos, y eventualmente manifestaciones de sobrecarga cardíaca y signos neurológicos de hipertensión endocraneana, incluyendo convulsiones y coma. Es propio de las últimas etapas de la gestación.
Síndrome de Eddowes. ver Síndrome de Digihton-Adair
Síndrome de Ehlers-Danlos. Síndrome asociado a alteraciones hereditarias del tejido conectivo. Se caracteriza por hiperelasticidad y fragilidad de la piel, laxitud articular, y facilidad para desarrollar hemorragias subcutáneas y hematomas musculares que posteriormente se calcifican. El trastorno se trasmite como un rasgo autosómico dominante.
Síndrome de Eisenmenger. Síndrome caracterizado por malformaciones cardíacas e hipertensión pulmonar. Se define por la presencia de una comunicación interventricular alta, con cabalgamiento de la aorta sobre el defecto septal, hipertrofia del ventrículo derecho y dilatación de la arteria pulmonar, que se evidencia por el agrandamiento del arco medio en las radiografías frontales del tórax.
Síndrome de la enfermedad del nódulo sinusal Síndrome de disfunción del nódulo sinusal de etiología desconocida. Clínicamente se manifiesta por accesos pasajeros de debilidad, fatiga fácil, mareos y a veces estados sincopales, que se repiten con una frecuencia variable en individuos sin signos de cardiopatía subyacente, y que obedecen a episodios súbitos de bradiarritmia (bradicardia sinusal, bloqueo sinoauricular, paro sinusal). El electrocardiograma de base suele ser normal, de modo que el diagnóstico solo puede establecerse accidentalmente (aparición de la anomalía durante la obtención de un registro) o si el médico lo sospecha y solicita un registro continuo prolongado con el método de Holter.
Síndrome de Erb. ver Síndrome de la miastenia grave.
Síndrome del escaleno anterior. ver Síndrome del estrecho superior del tórax.
Síndrome del esfuerzo. ver Síndrome de Da Costa.
Síndrome del espacio posteroexterno del cóndilo. ver Síndrome de Collet-Sicard.
Síndrome del espacio retroparotídeo. ver Síndrome de Villaret
Síndrome esquizofrénico. Síndrome psicótico caracterizado por una profunda desinserción del individuo de la realidad, a partir de severas alteraciones de la personalidad, el pensamiento y la afectividad. Se lo observa en la adolescencia y en la edad joven y se manifiesta, básicamente, por: a) trastornos de la personalidad, con pérdida del sentido de la individualidad y la singularidad, y a menudo con la convicción de estar controlado por personas o fuerzas, ajenas a él mismo; b) alteraciones del pensamiento, por las cuales el individuo explica su entorno sobre la base de hechos intrascendentes de esa realidad, a los que asigna un significado arbitrario, y de las construcciones racionales, rígidamente estructuradas, que hace a partir de esos hechos; c) oscurecimiento de las formas de expresión, que tornan crípticas y sólo comprensibles para él mismo; d) abulia, inercia, negativismo, estupor, y eventualmente, catatonía y autismo; e) pronunciada desafectivización y f) conservación de la inteligencia. En la práctica el síndrome tiene expresiones agudas (brotes esquizofrénicos) y variantes evolutivas crónicas (formas simples, hebefrénica, catatónica y paranoide).
Síndrome del estrecho superior del tórax. Síndrome neurovascular por irritación del plexo braquial. Es un complejo sintomático referido al miembro superior o a alguna de sus partes, en especial la mano, que se manifiesta por dolor, parestesias y fatigabilidad; por alteraciones vasomotoras: locales (acrocianosis, palidez, frialdad, fenómeno de Raynaud, y en ocasiones edema leve) y eventualmente por alteraciones tróficas (ulceraciones digitales) y atrofias musculares localizadas. Obedece a la irritación, compresión o estiramiento de los componentes del plexo braquial por estructuras vecinas, y engloba cuadros de distinto origen aunque de parecidas consecuencias : a) el síndrome del escaleno anterior, en el cual la arteria subclavia y el plexo braquial son comprimidos entre el escaleno anterior, la primera costilla y el escaleno mediano; b) el síndrome de la costilla cervical, donde la compresión se facilita por la presencia de una costilla rudimentaria nacida de la séptima vértebra cervical c) el síndrome costoclavicular originado en las estructuras, osteoligamentosas que unen la clavícula con la primera costilla, y d) el síndrome de hiperabducción, en el cual los trastornos no se producen por compresión, sino por estiramiento, y como consecuencia de posiciones anormales mantenidas durante un tiempo prolongado (p. ej., personas que duermen con una mano apoyada detrás de la cabeza o que desempeñan actividades laborales que requieren movimientos de hiperabducción).
Síndrome de exoftalmía maligna. Síndrome oftalmopático de la tirotoxicosis. Se manifiesta por una pronunciada exoftalmía bilateral, con paresia de los músculos oculomotores externos, tumefacción de los párpados, edema e inyección de la conjuntiva, y dolor retroocular constante. Es propio de la enfermedad de Graves.
Síndrome de Fanconi. Denominación general del conjunto de anomalías que resultan de una disfunción de los túbulos proximales. Se lo observa como forma idiopática o asociado a una variedad de procesos patológicos (amiloidosis, nefrosis, intoxicación por tetraciclinas caducas, etc.), y se lo define a partir de los siguientes criterios: a) por la presencia de cuatro anomalías básicas: glucosuria no asociada a hiperglucemia, fosfaturia, aminoaciduria y acidosis tubular renal; b) por las manifestaciones clínicas propias de estas anomalías que en general pueden ser manejadas satisfactoriamente pérdida de glucosa, formación solamente de cálculos de cistina, osteoporosis, acidosis metabólica, depleción de potasio,. etc. y c) por la ausencia de una cistinosis asociada.
Síndrome de Favre-Racouchout. Síndrome congénito caracterizado por alteraciones focales del tejido conectivo. Se manifiesta por la presencia de comedones y placas amarillentas circunscritas, engrosadas, que se ubican en la cara, alrededor de los ojos y de la nariz.
Síndrome de Felty. Síndrome de la artritis reumatoidea asociada a esplenomegalia y trastornos hematológicos. Se lo observa en pacientes con artritis reumatoidea de evolución prolongada e incluye esplenomegalia con leucopenia, anemia y trombocitopenia, y en ocasiones, fenómenos vasculíticos que originan úlceras cutáneas y neuropatía periférica. El mecanismo causal de estas alteraciones aún no se conoce.
Síndrome de feminización suprarrenal. Variedad del síndrome adrenogenital. Se manifiesta en el hombre por ginecomastia, atrofia testicular, cambios feminoides en la conformación corporal y en la implantación del vello, y aumento en la excreción urinaria de metabolitos estrogénicos. La causa más frecuente es un tumor suprarrenal secretor de androstenediona, que se convierte periféricamente en estrona y estradiol, o raramente, una deficiencia de la 3-b-ol-deshidrogenasa, que desvía el metabolismo de la androstenediona hacia el de los progestágenos.
Síndrome de feminización testicular. Síndrome de seudohermafroditismo en individuos genéticamente masculinos. Se caracteriza por la presencia de testículos, feminización total de los genitales externos y de la conformación corporal, y ausencia o hipoplasia acentuada del útero y de las trompas, con niveles normales de testosterona circulante. Se deben a una resistencia a la testosterona de los órganos efectores.
Síndrome fetal alcohólico. ver Síndrome alcohólico fetal
Síndrome de Fitz-Hugh-Curtis Síndrome de perihepatitis por distintas infecciones abdominopelvianas. Se manifiesta por fiebre, dolor y contractura de los músculos de la pared abdominal, dolor más intenso en el cuadrante superior derecho, y ocasionalmente ruido de fricción auscultable a nivel del hipocondrio derecho. La causa más; común, es la gonococia; aunque también se lo ha observado en salpingitis por clamidias y quizá por otros agentes causales.
Síndrome de la flexura esplénica. Síndrome seudoanginoso de origen digestivo. Incluye dolorimiento y molestias., inespecíficas en el cuadrante superior izquierdo del abdomen, que puede originar dolor en la región precordial y en el hombro y brazo izquierdos. Su causa habitual es el meteorismo colónico.
Síndrome de Foix. Síndrome neurológico caracterizada por compromiso de los nervios craneanos V, VII, IX, X y XII, Y por profrusión de un globo ocular. Se manifiesta por oftalmoplejía unilateral completa (nervios craneanos m, IV Y VI) asociada a anestesia de la ·córnea (rama oftálmica del V nervio), con el agregado· de exoftalmos homolateral, lo cual lo distingue del síndrome de Rochon-Duvigneau. Es producido por tumores y aneurismas del seno cavernoso, o por tumores invasivos de los senos venosos o de la silla turca.
Síndrome de Foix-Alajouanine. Síndrome neurológico de la mielitis necrosante subagudo. Se manifiesta por una paraplejía dolorosa que primero es espástica y luego se hace fláccida, con atrofia muscular, arreflexia tendinosa, anestesia e incontinencia esfinteriana. La causa parece ser degenerativa, secundaria a procesos infecciosos o de origen vascular.
Síndrome de Forbes-Albright. Síndrome endocrino por hipersecreción de prolactina. Se caracteriza por galactorrea sostenida en mujeres que no se encuentran en periodo puerperal, acompañada de niveles elevados de prolactina circulante. Es causado por tumores de la adenohipófisis y, con menos frecuencia, del hipotálamo.
Síndrome de Foster-Kennedy. Síndrome de anosmia unilateral de causa orgánica. Se manifiesta únicamente por anosmia unilateral y es producido por aneurismas arteriales o, más a menudo, por tumores de la base del lóbulo frontal que comprimen el bulbo y la cintilla olfatoria.
Síndrome de Frey. Síndrome irritativo de ciertas afecciones parotídeas. Se caracteriza por enrojecimiento, sudoración y sensación de calor en el territorio del nervio auriculotemporal, desencadenados por la masticación. Se lo observa en lesiones unilaterales de la parótida (tumores, supuraciones) o del simpático cervical.
Síndrome de Froin. Síndrome relacionado con alteraciones del líquido cefalorraquídeo de causa no infecciosa. Define al líquido cefalorraquídeo que es xantocrómico (color amarillo claro, trasparente), con gran contenido proteico, rápida coagulación y cantidad normal de células. Estas características se observan en casos de interrupción del flujo entre los ventrículos cerebrales y las zonas más distales.
Síndrome de la galactorrea por drogas. Síndrome endocrino por hipersecreción de prolactina. Se caracteriza por galactorrea en la mujer, y por turgencia mamilar o ginecomastia en el hombre, que aparecen durante el tratamiento con ciertos medicamentos (entre ellos la espironolactona, la fenotiazina y otros tranquilizantes y antidepresivos, la reserpina, la metildopa, los digitálicos, etc.) y que desaparecen cuando se interrumpe su administración.
Síndrome del ganglio geniculado. ver Síndrome de Ramsay-Hunt
Síndrome de Ganser Síndrome de ciertas alteraciones del comportamiento en pacientes con histeria. Se manifiesta en una personalidad histérica, por amnesia, alteraciones atípicas de la conciencia, actos absurdos y respuestas sin sentido a las preguntas.
Síndrome de Garcin. Síndrome neurológico de la base del cráneo. Parálisis unilateral de todos los pares craneanos, especialmente de los diez últimos, sin signos de hipertensión endocraneana. Es causado por tumores extensos o metástasis tumorales que infiltran progresivamente la base del cráneo.
Síndrome de Gardner. Síndrome hereditario caracterizado por alteraciones cutáneas y tendencia al desarrollo de procesos malignos. Se manifiesta por la presencia de tumores dermoides, lipomas, fibromas, osteomas y quistes epidérmicos y sebáceos, asociados a una poliposis colónica. La incidencia de adenocarcinomas del colon es sumamente elevada, a menos que el individuo sea colectomizado. El cuadro se trasmite como un rasgo autosómico dominante.
Síndrome gastrocardíaco. Síndrome funcional del corazón de origen gástrico. Se manifiesta por extrasístoles, episodios transitorios de taquicardia o cuadros seudoanginosos, sin relación con el esfuerzo y que habitualmente aparecen en el período posprandial. Se los atribuye a compresión de la cámara gástrica contra la pared inferior del diafragma y con cierta frecuencia existe una hernia hiatal asociada.
Síndrome de Gélineau. Síndrome de la narcolepsia. En su forma completa se caracteriza por la siguiente tétrada: a) ataques recurrentes, irresistibles, de somnolencia diurna (narcolepsia); b) episodios breves, bruscos, de pérdida del tono muscular sin pérdida de la conciencia ( cataplexia); c) sensación atemorizante de incapacidad de mover los músculos voluntarios al comienzo del sueño o en el despertar (parálisis del sueño), y d) alucinaciones al iniciar el sueño (hipnagógicas) o al despertar (hipnopómpicas ) La causa aún no ha sido aclarada.
Síndrome general de adaptación. Síndrome de la respuesta global del organismo a las situaciones de tensión. Conjunto de reacciones sistémicas consecutivas a la exposición sostenida a condiciones de stress (traumatismos, frío, calor, fatiga, etc.). Comprende tres estadios: reacción de alarma, estadio de resistencia y estadio de agotamiento.
Síndrome de Gerstrnann. Síndrome neurológico por lesión del giro angular dominante. Combinación de agnosia táctil, dificultad para distinguir entre la derecha y la izquierda, agrafia, acalculia, y a menudo, apraxia de prensión. Se lo observa en las lesiones del giro angular izquierdo, entre la corteza parietal y la occipital, y por lo general es de origen tumoral.
Síndrome de Gilles de la Tourette. Síndrome de tics múltiples de evolución crónica. Se caracterizan por tics faciales que aparecen en la infancia y que progresan engendrando sacudidas: amplias; estereotipadas, en el resto del organismo, y que típicamente se acompañan de ecolalia y coprolalia. La causa se atribuye a una combinación de factores genéticos y ambientales y el trastorno persiste durante toda la vida.
Síndrome de Goodpasture. Síndrome de la glomerulonefritis asociada a alteraciones pulmonares y hemoptisis. El punto de partida es un cuadro respiratorio con hemoptisis e infiltrados pulmonares difusos, bilaterales, seguido de anemia y de una glomerulonefritis rápidamente evolutiva que se manifiesta por hematuria, proteinuria intensa, hipertensión arterial e insuficiencia renal. El desenlace suele ser fatal y se lo atribuye a fenómenos de Antoinmunidad.
Síndrome de Gorlin-Goltz. Síndrome hereditario caracterizado por alteraciones cutáneas y tendencia al desarrollo de procesos malignos. Se define por la presencia de carcinomas basocelulares múltiples, quistes epidermoides y hoyuelos en las palmas y las plantas; además de fibromas ováricos y quistes maxilares, y en cierta proporción de casos, de hipertelorismo y anomalías de los metacarpianos. Se trasmite como un rasgo autosómico dominante y se ha demostrado una tendencia definida al desarrollo de fibrosarcomas del maxilar inferior y meduloblastomas.
Síndrome de Gougerot-Carteaud. Síndrome de la papilomatosis confluente y reticulada. Se caracteriza por la presencia de gran cantidad de pápulas discretas; que luego confluyen para convertirse en pápulas verrugosas. Se localiza en la línea media del tronco y pliegues de flexión del codo e involuciona lentamente. Suele afectar a niñas en edad puberal.
Síndrome de Gradenlgo. Síndrome neurológico con compromiso de los nervios craneanos V y VI. Se define por la presencia de parálisis unilateral de la desviación de la mirada hacia afuera (lesión del VI nervio craneano) acompañada de cefalea y neuralgia facial homolateral (lesión del V nervio craneano). Se lo observa en procesos supurativos del oído, medio y en los tumores de la fosa del peñasco.
Síndrome de Groenblad-Straudberg. Síndrome del seudoxantoma elástico. Sus rasgos distintivos son: a) en la piel, aparición de zonas romboidales degenerativas, de atrofia y flaccidez, que predominan en el cuello, la axila y la ingle y se desarrollan a partir de los 30-40 años; b) disminución progresiva de la visión, relacionada con la presencia de bandas angioideas, de un color pardo o pardo grisáceo en la superficie de la retícula, y c) signos crecientes de insuficiencia circulatoria periférica producidos por calcificación y oclusión de arterias de mediano calibre, incluyendo las del miembro superior. La afección es de causa desconocida y se trasmite como un rasgo autosómico recesivo.
Síndrome de Guillaln-Barré. Síndrome de la polineuritis idiopática aguda: Debilitamiento rápidamente progresivo de las neuronas motrices ascendentes que suele presentarse después de una infección respiratoria o entérica. Se inicia con parestesias de los pies, seguidas de parálisis fláccida y debilidad de las piernas; que ascienden a los brazos, tronco y cara; se acompaña de fiebre baja, parálisis bulbar, ausencia o disminución de los reflejos tendinosos y aumento de las proteínas del líquido cefalorraquídeo sin aumento celular concomitante. Se piensa que la causa es inmunológica y en el 75% de los pacientes la recuperación es completa.
|
|
Síndrome de Gunn. Síndrome relacionado con movimientos asociados de un párpado y del maxilar inferior. Se caracteriza por ptosis palpebral unilateral, casi siempre congénita, y exagerada elevación del párpado ptosado al mover la mandíbula en sentido contralateral; en ciertos casos también ocurre lo inverso: la estimulación de la córnea suscita movimientos de la mandíbula hacia el lado contrario. La causa se desconoce y no existen otros trastornos corporales.
Síndrome de Hallopeau-Siemens. Síndrome de la epidermólisis ampollar polidisplásica. Consiste en ampollas que se desarrollan desde épocas tempranas de la vida y que curan dejando cicatrices, sobre las que pueden aparecer epiteliomas. Las mucosas están afectadas con frecuencia. Es un trastorno hereditario recesivo.
Síndrome de los hamartomas múltiples. Síndrome hereditario caracterizado por alteraciones cutáneas y tendencia al desarrollo de procesos malignos. Se manifiesta por la presencia de: a) pápulas verrugosas en la piel; b) fibromas en la mucosa bucal; c) hamartomas múltiples en otras regiones del organismo (fibroquistosis mamaria, neuromas, adenomas tiroideos, lipomas y hemangiomas), y d) tendencia aumentada al desarrollo de carcinomas. mamarios y de la glándula tiroides. El trastorno es trasmitido como un rasgo autosómico dominante.
Síndrome de Hamman-Rich. Síndrome de la fibrosis pulmonar idiopática. Se caracteriza por el desarrollo de tejido fibroso en el intersticio pulmonar, de evolución crónica, subaguda o aguda. Existe disnea progresiva y luego permanente, hipocratismo digital y alteración en el trasporte de gases a través de la pared alveolar. Es irreversible y de causa desconocida.
Síndrome de Harris. Síndrome del hiperinsulinismo endógeno. Se manifiesta por episodios más o menos frecuentes de desasosiego, palidez, traspiración, taquicardia, confusión mental y alteraciones de la visión, relacionados con una disminución marcada de los niveles plasmáticos de glucosa. Es producido por hipersecreción de insulina a partir de tumores o hiperplasias difusas de las células beta de los islotes de Langerhans.
Síndrome de Hedblom. Síndrome de la inflamación aguda del diafragma. Se manifiesta por dolor inspiratorio, inmovilidad de la parte inferior del tórax durante la inspiración y dolor bilateral en la porción superior del abdomen, sin patología abdominal que justifique el cuadro. Se debe a una miositis aguda primaria del diafragma.
Síndrome de Heerfordt. Síndrome de la fiebre uveoparotídea. Se caracteriza por inflamación uveal y tumefacción parotídea, asociadas a parálisis, facial bilateral (diplejía facial) con o sin parálisis de otros nervios craneanos. Es una de las variantes de iniciación de la sarcoidosis.
Síndrome de Heidenhain. Síndrome neurológico de una demencia presenil. Se trata de un cuadro demencial rápidamente evolutivo acompañado de ceguera cortical, disartria, ataxia, atetosis y rigidez generalizada, sin alteraciones en el fondo del ojo. El trastorno es degenerativo y de causa desconocida.
Síndrome del hemangioma con trombocitopenia. ver Síndrome de Kasabach-Merritt.
Síndrome del hematoma subdural. Síndrome neurológico de las colecciones venosas intracraneales. Se manifiesta por cefalea, somnolencia, confusión mental, signos motores focales (monoparesias, hemiparesia), asimetría pupilar, edema de papila, y eventualmente estupor, coma y convulsiones c1ónicas. Suele ser de progresión insidiosa, solapada, y es producido por la expansión lenta de un hematoma intracraneal de origen venoso, en pacientes con o sin antecedentes llamativos de un traumatismo de cráneo, de corta o mediana data (desde pocos días hasta uno o más meses)
Síndrome de la hemianopsia bitemporal. Síndrome neurooftalmológico de la lesión bilateral de los nervios ópticos. El cuadro es causado por adenomas de la hipófisis, meningiomas de la silla turca o aneurismas saculares del polígono de Willis, que al expandirse comprimen simultáneamente la porción interna de ambos nervios ópticos, es decir, las fibras que se dirigen a la mitad nasal de ambas retinas. Como consecuencia de esto se produce una ceguera para los objetos situados en el lado temporal de ambos campos visuales (hemianopsia bitemporal).
Síndrome de la hendidura esfenoidal. ver Síndrome de Rochon-Divigneau.
Síndrome de la hiperabducción. ver Síndrome del estrecho superior del tórax.
Síndrome de la hiperpotasemia. Síndrome muscular y electrocardiográfico de la hiperpotasemia. Se hace evidente cuando los niveles de potasio sérico superan los 6 mEq/l, y se caracteriza por debilidad muscular creciente, hiporreflexia o arreflexia osteotendinosa, y alteraciones típicas en el electrocardiograma: primero aparecen ondas T puntiagudas en todas las derivaciones y luego, sucesivamente, alargamiento del P-R, desaparición de la onda P, alteraciones del complejo QRS, y eventualmente fibrilación ventricular y paro cardíaco. Se lo observa en la enfermedad de Addison, en las fases terminales de la insuficiencia renal crónica, y fundamentalmente en las formas. hipercatabólicas de la insuficiencia renal aguda.
Síndrome de la hiperqueratosis palmoplantar. Síndrome hereditario con alteraciones cutáneas y tendencia al desarrollo de procesos malignos. Se caracteriza por la presencia de hiperqueratosis de la palma de las manos y la planta de los pies, que comienza ya en la segunda infancia, sin otras anomalías: detectables. La incidencia posterior de carcinomas del esófago es sumamente elevada, y la afección se trasmite como un rasgo autosómico dominante.
Síndrome hipercinético de la infancia. Síndrome de aceleración de las funciones psicomotoras en el niño. Se caracteriza por hiperactividad motora, impulsividad, inquietud, marcada incapacidad de fijar la atención y escasa tolerancia a la frustración. La causa no se conoce, si bien en la mayoría de los casos no es orgánica, y el cuadro suele desaparecer en la adolescencia.
Síndrome de hipersomnia con apnea nocturna. Síndrome apneico asociado a cuadros de obesidad de distinta etiología. El cuadro típico es el de una hipersomnia diurna con pausas; ventilatorias de frecuencia y duración anormal que se producen durante el sueño. Se lo observa en el síndrome de Pickwick, en la obesidad simple, en el mixedema y en otros estados patológicos.
Síndrome de hipertensión portal. Síndrome de hipertensión venosa por obstrucción del sistema porta. Se manifiesta por ascitis y edema periférico, esplenomegalia congestiva, desarrollo de circulación venosa colateral (abdomen y pared anterior del tórax), dilatación de otros sistemas venosos (várices esofágicas), oliguria, y eventualmente hemorragias digestivas (rotura de várices esofágicas). Se lo observa como manifestación primaria en la pileflebitis de la vena porta, y secundariamente en la cirrosis hepática y en otros procesos obstructivos.
Síndrome de hiperventilación. Síndrome de la hiperventilación pulmonar de causa no orgánica. Es un cuadro de angustia y sensación de falta de aire, entumecimiento y parestesias en la cara y en los miembros, y calambres en las manos y los pies. Habitualmente se produce durante la noche, cuando el individuo intenta conciliar el sueño o a poco de haberse dormido. La causa es una hiperventilación desencadenada por factores emocionales.
Síndrome de la hipopotasemia. Síndrome muscular y electrocardiográfico de la hipopotasemia. Se caracteriza por debilidad general que incluso puede llegar a la parálisis completa. (como se observa en la parálisis periódica familiar), acompañada de anomalías electrocardiográficas típicas: aplanamiento generalizado de las ondas: T, ondas U prominentes, disminución del ST, y eventualmente paro cardíaco en asistolia. La causa más frecuente es la ingestión de diuréticos, por automedicación o por prescripción médica y también puede producirse por abuso crónico de laxantes, en los cuadros de vómitos abundantes o diarreas profusas, etcétera.
Síndrome de hipotensión endocraneana. Síndrome neurológico de la hipotensión del líquido cefalorraquídeo. Se manifiesta por náuseas, vértigo y acufenos, bradicardia, cefalea que alivia en decúbito, rigidez de nuca, y edema de papila bilateral de leve a moderado. Es complicación relativamente frecuente de los traumatismos de cráneo y se debe a una interrupción de la formación de líquido cefalorraquídeo con la consiguiente tendencia al colapso de los espacios ventriculares.
Síndrome de hipotensión ortostática idiopática. Síndrome asociado a la degeneración periférica o central de estructuras del sistema nervioso autónomo. Se caracteriza por una hipotensión ortostática cuya severidad puede ser tal como para causar síncope o accesos convulsivos. El cuadro es progresivo, se observa en personas de edad y se acompaña de anhidrosis ascendente, disminución del metabolismo basal y de la producción de norepinefrina, deficiente secreción de lágrimas y saliva, atonía vesical y otras manifestaciones.
Síndrome de hipoventilación idiopática. Síndrome de hipoventilación con manifestaciones clínicas, voluntariamente corregible, y de causa desconocida. Es un cuadro raro que afecta a individuos sin patología pulmonar ni de la pared torácica, sin alteraciones en las pruebas funcionales respiratorias, y sin anomalías detectables en el sistema nervioso central. Se caracteriza: a) por una tendencia sostenida involuntaria e insensible a hipoventilar, suficientemente importante como para que se eleve la PC02 y disminuya la P02 de la sangre, y para que el individuo se encuentre en un estado de permanente debilidad, cansancio y somnolencia, y b) por la capacidad del individuo de revertir, la situación en la medida en que advierte, o que se le advierte lo que le está ocurriendo, e intensifica voluntariamente la actividad ventilatoria. Los valores del hematócrito son superiores al 50% y en la mayoría de los casos se observan episodios de respiración periódica durante el sueño.
Síndrome de Hippel-Landau. Síndrome neurooftalmológico asociado a malformaciones vasculares congénitas. Anatómicamente, se define por la presencia simultánea de hemangiomas capilares múltiples en la retina, y de uno o más hemangioblastomas en el cerebelo, de crecimiento lento y con formaciones quísticas. Y clínicamente, por cefalea y edema de papila, y manifestaciones progresivas de ataxia cerebelosa y de pérdida de la visión. Existen casos de aparición espontánea y otros que se trasmiten como un rasgo autosómico dominante.
Síndrome de Holt-Oram. Síndrome asociado a mal formaciones cardiovasculares y del sistema esquelético. Asociación de defectos del tabique interauricular, u otras anomalías, cardíacas, con hipoplasia de las clavículas y malformaciones de los miembros superiores.
Síndrome de Holzknecht-Jacobson. Síndrome radiológico de la atelectasia pulmonar. Se caracteriza por opacidad del campo pulmonar afectado, desviación del mediastino hacia ese lado; elevación del diafragma homolateral y retracción de los espacios intercostales.
Síndrome del hombre rígido. ver Síndrome Wolt Man-Moersch
Síndrome hombro-mano. ver Síndrome de la distrofia simpática refleja.
Síndrome de Homen. Síndrome neurológico de la degeneración del núcleo lenticular. Es una afección progresiva e irreversible que se caracteriza por vértigo disartria, ataxia, rigidez que predomina en los miembros inferiores y demencia. El origen es genético.
Síndrome de Horner. ver Síndrome de Claude Bernard Horner.
Síndrome de Horton. Síndrome de la cefalea histamínica Se caracteriza por ataques de dolor unilateral sobre el ojo y la frente, de comienzo y terminación bruscos; hay enrojecimiento, calor y sudoración del lado afectado, por dilatación de la arteria temporal. Predomina en hombres, durante el sueño, y se atribuye a una descarga local de histamina
Síndrome de Hunt. Síndrome de la parálisis agitante juvenil. Se manifiesta desde los primeros años de vida por hipertonía muscular y facies y actitud general propias de la enfermedad de Parkinson. Es familiar y obedece a la degeneración progresiva del globo pálido y otros núcleos vecinos.
Síndrome de Hunter. Síndrome de la mucopolisacaridosis II. Se caracteriza por: a) escafocefalia con cuello corto, cejas hirsutas, lengua grande y prominencia lateral de las paredes externas de las órbitas; b) rigidez articular, con pies cavos y manos en flexión; c) ausencia de opacidades corneales, y d) sordera, pero con retardo mental menos severo que el del síndrome de Hurler. Se lo observa únicamente en varones, que pueden alcanzar la edad media de la vida, y obedece a una deficiencia de iduronato sulfatasa, trasmitida como un rasgo recesivo ligado a X.
Síndrome de Hurler. Síndrome de la mucopolisacaridosis 1H. Sus rasgos básicos son: a) cabeza voluminosa con escafocefalia, cejas hirsutas; frente y ojos prominentes, lengua gruesa y mejillas abultadas (gargoilismo, por la similitud con las gárgolas de la arquitectura gótica); b) estatura, cuello y extremidades cortas, con cifosis acentuada y vientre prominente; c) opacidades corneales, y d) sordera y retardo mental severo. El trastorno es genético, obedece a una deficiencia de a-L-iduronidasa y la muerte suele producirse antes de los 10 años
Síndrome de incontinencia pigmentaria. ver Síndrome de Dloch-Sijlzberger.
Síndrome de inmunodeficiencia combinada severa. Síndrome congénito relacionado con una insuficiencia global de los mecanismos inmunitarios. Se caracteriza por trastornos graves de la inmunidad humoral y celular, con deficiencia de inmunoglobulinas y linfopenia marcada que incluye las células, T y B. Hay infecciones repetidas y los pacientes, por lo general, mueren durante el primer año de vida. El trastorno es de tipo congénito hereditario o aparece bajo la forma de casos esporádicos.
Síndrome de inmunodeficiencia con timoma. Síndrome de deficiencia inmunológica de causa no determinada Se observa en pacientes con timomas de células fusiformes y se caracteriza por ausencia o disminución marcada de linfocitos B en la circulación y de sus precursores en la médula ósea, juntamente con eosinopenia, niveles bajos de inmunoglobulinas plasmáticas, y en ocasiones, aplasia eritroide.
Síndrome de insuficiencia poliglandular. Síndrome de la insuficiencia simultánea de dos o más sistemas endocrinos. El síndrome es producido por distintos cuadros de hipofunción endocrina (insuficiencia suprarrenal, hipoparatiroidismo, tiroiditis linfocítica, insuficiencia gonadal) y por las manifestaciones que resultan de la combinación de dos o más de ellos entre sí, a los cuales a menudo se asocia una diabetes y enanismo. No se detectan anomalías significativas en la adenohipófisis y sus rasgos básicos son los bajos niveles de hormonas circulantes y la existencia de anticuerpos contra dos o más sistemas endocrinos.
Síndrome de insuficiencia respiratoria del recién nacido. Síndrome de insuficiencia respiratoria aguda de etiología múltiple. Sus dos signos fundamentales son la disnea y una acentuada taquipnea (más; de 60 respiraciones por minuto), acompañada de grados variables de taquicardia, cianosis y alteraciones en el estado acidobase. Se produce por causas: centrales (depresión medicamentosa, inmadurez o anoxia cerebral etc.) o periféricas: procesos infecciosos, aspiración de líquido amniótico, atelectasias, inmadurez pulmonar (síndrome de Wilson-Mikity), depósito de material hialino en los alvéolos y bronquíolos (síndrome de la membrana hialina), etcétera.
Síndrome interolivar. ver Síndrome de Dejerine.
Síndrome de la intolerancia a la fructosa. ver Síndrome de deficiencia de aldolasa hepática.
Síndrome de intoxicación digitálica. Síndrome producido por la ingestión de dosis excesivas de digitálicos. Incluye trastornos digestivos, que suelen ser los primeros en aparecer (anorexia acentuada, estado nauseoso persistente), alteraciones visuales (visión borrosa o coloreada, escotomas) y anomalías electrocardiográficas cuya gravedad suele ser proporcional a la intensidad de la intoxicación aparecen arritmias del tipo de la extrasistolia aislada, el bigeminismo extrasistólico o la extrasistolia ventricular multifocal, seguidas de bloqueo auriculoventricular de primero o segundo grado, y eventualmente, de un bloqueo, completo.
Síndrome por irradiación. Síndrome producido por la sobreexposición aguda a los rayos roentgen. Se manifiesta por anorexia, debilidad y fiebre, sed intensa y postración, seguidas al cabo de poco tiempo de anemia, granulocitopenia, pérdida de peso, caída de cabello, y alteraciones generalizadas que en los casos de irradiación masiva (escapes radiactivos, explosiones nucleares, etc.) conducen rápidamente a la muerte del individuo.
Síndrome de Ivemark. Síndrome de los cuadros de agenesia del bazo con anomalías cardíacas y viscerales. Se caracteriza por agenesia del bazo, malformaciones cardíacas: habitualmente con cortocircuito de derecha a izquierda, e inversión de las vísceras de grado variable. El cuadro es congénito y obedece a alteraciones del desarrollo que se producen alrededor de la cuarta o quinta semana de la vida intrauterina.
 Síndrome de Jaccoud Síndrome de Jaccoud |
Síndrome de Jaccoud. Síndrome de una poliartritis crónica progresiva de evolución atípica. Define una poliartritis crónica progresiva de consecuencias virtualmente equiparables a las de la artritis reumatoidea (fibrosis periarticular, rigideces, desviación cubital de la mano, etc.) pero que no se inicia como tal, sino como una variante evolutiva en un individuo que era portador, en un principio, de las manifestaciones típicas reversibles del reumatismo poliarticular agudo (fiebre reumática).
Síndrome de Jackson. Síndrome neurológico por lesión del tronco cerebral con compromiso de los nervios craneanos IX y X. Se manifiesta por parálisis de la mitad de la lengua del lado de la lesión (IX nervio) más los signos propios del síndrome de Avellis. Es causado por tumores o focos de reblandecimiento a nivel del techo del bulbo y de la vía corticoespinal.
Síndrome de Jacod. Síndrome neurooftálmico por lesión de nervios craneales. Es unilateral y se caracteriza por neuralgia en el territorio del trigémino, ceguera y oftalmoplejía completa. Lo producen los tumores nasofaríngeos que se extienden a la encrucijada petroesfenoidal y afectan los nervios craneales II, III, V y VI.
Síndrome de Jaddasohn-Lewandowsky. Síndrome de la paquioniquia congénita. Se caracteriza por engrosamiento de las uñas; onicogriposis, hiperqueratosis de rodillas, codos, palmas y plantas, leucoplasia oral e hiperhidrosis de manos y pies. Se hereda como rasgo autosómico dominante.
Síndrome de Jackson-Wartenhorst. Síndrome de la policondritis recidivante. Es un proceso inflamatorio febril, doloroso, de los cartílagos de las orejas y la nariz, que conduce a la atrofia y puede afectar los cartílagos de las vías respiratorias y de las costillas. Se acompaña de artritis, episcleritis, iritis, miocarditis e insuficiencia aórtica. La causa se desconoce.
Síndrome de Jervell y Lange-Nielsen. ver Síndrome QT.
Síndrome de Kartagener. Síndrome de un cuadro de malformaciones cardiovasculares y de las vías respiratorias. Se define por la presencia de dextrocardia, situs inversus y bronquiectasias y por una tendencia a desarrollar infecciones pu1monares y sinusales recurrentes. El trastorno es hereditario y se trasmite como un rasgo autosómico dominante.
Síndrome de Kasabach-Merritt. Síndrome trombocitopénico relacionado con la presencia de un hemangioma, Se manifiesta por trombocitopenia marcada en pacientes portadores de un hemangioma gigante, sin alteraciones significativas en la médula ósea salvo un exceso de megacariocitos. El cuadro es propio del recién nacido y se normaliza con la extirpación del hemangioma.
Síndrome de Kennedy. Síndrome neurooftalmológico de origen tumoral. Se caracteriza por ceguera unilateral y edema de papila del lado opuesto. Lo producen los tumores del lóbulo frontal, o los meningiomas de la cresta esfenoidal, que comprimen progresivamente un nervio óptico (ceguera homolateral) y obstruyen el retorno venoso del lado opuesto (edema de papila contralateral).
Síndrome de Kimmelstiel-Wilson. Síndrome de la diabetes sacarina asociado a hipertensión y lesiones renales degenerativas. Estas últimas consisten en una glomerulohialinosis intercapilar y se manifiestan por hematuria, albuminuria y cilindruria, un síndrome nefrótico e insuficiencia renal progresiva.
Síndrome de Kinsbourne. Síndrome de la epilepsia mioclónicas de la infancia. Se caracteriza por contracciones mioclónicas del tronco, extremidades y cara, con ataxia dinámica y temblor intencional. La causa es desconocida, aunque en un 50% de los casos existe un neuroblastoma oculto.
Síndrome de Kleine-Levin: Síndrome de la hipersomnia periódica. Se caracteriza por periodos de hipersomnolencia que duran de días a semanas alternando con períodos asintomáticos de meses a años. Durante los primeros el individuo tiene un apetito voraz y puede manifestar alteraciones del ánimo y de la conducta sexual (hiperactividad, exhibicionismo) junto con alucinaciones, desorientación y trastornos de la memoria. Suele iniciarse en la juventud y la causa es desconocida.
Síndrome de Klinefelter. Síndrome de causa genérica caracterizado por alteraciones endocrinas y anomalías genitales. Se distingue por el aspecto eunucoide o débilmente masculinizado del paciente, ginecomastia, testículos pequeños e hipoplásicos, hialinización de los túbulos seminíferos con espermatogénesis y conservación de las células de Leydig, niveles elevados de gonadotrofinas urinarias (FSH) y secreción disminuida de andrógenos. El sexo genético suele ser femenino, existen tres o más juegos de cromosomas sexuales (XXY, XXXY o XXYY, XXXXY) y los cromosomas autosómicos son normales.
Síndrome de Klippel-Feil. Síndrome producido por la ausencia congénita de vértebras cervicales. Sus rasgos básicos son: a) acortamiento marcado o virtual ausencia del cuello, de modo tal que la cabeza parece apoyar directamente sobre la porción superior del tronco; b) incapacidad de girar la cabeza; c) implantación sumamente baja del cabello de la nuca, y d) en las radiografías se observa disminución del número de vértebras cervicales, consolidación de las existentes en un bloque único, y apertura de los arcos vertebrales posteriores (espina bífida).
Síndrome de Klippel-Trenaunay-Weber. Síndrome asociado a malformaciones unilaterales de origen congénito. El cuadro suele afectar a una extremidad y se caracteriza por crecimiento exagerado del miembro debido a hipertrofia ósea y de los tejidos blandos, a menudo con sindactilia u otras, malformaciones, y por la coexistencia de anomalías vasculares del tipo de los hemangiomas cutáneos, las dilataciones varicosas o los nevos en llama. La causa aún no ha sido establecida.
Síndrome de Konig. Síndrome de un cuadro de dolor intermitente en la fosa ilíaca derecha con alteraciones del tránsito intestinal. Se caracteriza por la aparición intermitente de manifestaciones a nivel de la fosa ilíaca derecha (dolor cólico y distensión localizada, seguidos de borborigmos que alivian rápidamente la sintomatología) y por períodos de constipación que alternan con otros de diarrea. Se lo observa en procesos inflamatorios o neoplásicos que afectan al ciego o a las zonas vecinas.
Síndrome de Korsakoff. Síndrome de la psicosis amnésica confabulatoria. Se la observa en alcohólicos crónicos y se caracteriza por trastornos de la orientación, susceptibilidad a estímulos externos y a la sugestión, alucinaciones, confabulación y fundamentalmente amnesia, que es desproporcionada en relación con las restantes manifestaciones mentales y del comportamiento. Suelen existir alteraciones polineuríticas (mano péndula, etc.).
Síndrome de Koshevnicoff. Síndrome de ciertas formas graves de la epilepsia motora. Incluye convulsiones repetidas, hipertemia, delirio, pérdida de la conciencia y parálisis de distribución variable, junto con postración y persistencia de movimientos mioclónicos en los períodos intercríticos.
Síndrome del kwashiorkor. Síndrome de la desnutrición caloricoproteica grave de la infancia. Se observa en niños pequeños y se caracteriza por anorexia, edemas e hipoalbuminemia marcada, alteraciones cutáneas (despigmentación con manchas eritematosas, dermatosis. varías), descoloración en bandas del cabello, aspecto misérrimo y falta de respuesta emocional. Obedece a deficiencias, alimentarias múltiples y prolongadas.
Síndrome de las lágrimas de cocodrilo. ver Síndrome de Bogorad.
Síndrome de Landry-Guillain-Barré. ver Síndrome de Guillain-Barre.
Síndrome de Laubry-Soule. Síndrome digestivo asociado al infarto de miocardio. Se lo observa en algunos pacientes con infarto de miocardio y se manifiesta por meteorismo localizado a nivel del ángulo izquierdo del colon y de la cámara gástrica. Se lo considera un fenómeno reflejo.
Síndrome de Launois. Síndrome que acompaña a ciertas formas de hiperpituitarismo. Es un cuadro de gigantismo que se inicia antes del cierre de las epífisis, habitualmente en la pubertad, y que resulta de una hipersecreción de hormonas hipofisarias de origen tumoral (adenomas eosinófilos, y en ocasiones adenomas, cromófobos).
Síndrome de Laurence-Moon-Biedl. Síndrome hereditario producido por disfunción hipotalámica. Sus componentes son los del síndrome adiposogenital de Frohlich (obesidad feminoide, infantilismo sexual, retardo mental, etc.), a los que se agregan retinitis pigmentaria y polidactilia. No hay alteraciones en la adenohipófisis y el cuadro es de origen genético.
Síndrome de leche y álcalis. Síndrome de hipercalcemia por ingestión prolongada de álcalis absorbibles. Sus manifestaciones incluyen prurito, debilidad muscular y dolores reumatiformes, polidipsia y poliuria, hipercalcemia sin hipercalciuria, y calcinosis generalizada que eventualmente puede conducir a la insuficiencia renal. El cuadro es propio de ulcerosos que consumen grandes cantidades de leche y alcalinos, y su incidencia ha disminuido notablemente desde la introducción de los antiácidos no absorbibles.
Síndrome de Leriche. Síndrome de la obstrucción crónica de la porción terminal de la aorta abdominal. Se caracteriza por palidez y enfriamiento, de los miembros inferiores, debilidad de los músculos de la cadera, las piernas o los tobillos durante la marcha o el ejercicio, impotencia sexual y ausencia de los pulsos femorales.
Síndrome de Lermoyez. Síndrome de ciertas alteraciones transitorias de la función auditiva y vestibular. Incluye tinnitus y sordera de aparición repentina, que preceden a una crisis de vértigo y se atenúan una vez establecido éste. Se lo atribuye a espasmos sostenidos de la arteria auditiva interna.
Síndrome de Lewis. Síndrome hereditario debido a anomalías congénitas en el metabolismo de los carbohidratos. Incluye retardo mental de grado variable, trastornos neurológicos (en particular convulsiones) y crisis hipoglucémicas que predominan en horas de la noche. La causa es una deficiencia congénita de la glucogenosintetasa hepática.
Síndrome de Lichtheim. Síndrome de la degeneración combinada subaguda de la médula espinal. Se manifiesta por parestesias; ataxia, inestabilidad en la marcha y luego paraplejía espástica. Se lo observa en la anemia perniciosa idiopática o de Biermer y obedece a una degeneración de las columnas laterales y posteriores de la médula espinal
Síndrome de Liddle. Síndrome del seudohiperaldoseronismo. Se caracteriza por hipertensión y alcalosis hipoc1orémica, con secreción insignificante de aldosterona. Es hereditario, probablemente de trasmisión autosómica dominante, y la causa se desconoce.
Síndrome de Lightwood. Síndrome de la incapacidad del riñón de excretar orina suficientemente ácida. Se manifiesta por una acidosis metabólica persistente con hipercloremia, y eventualmente con depleción de potasio e hipercalciuria. Obedece a un defecto primario de la función de los túbulos renales (de tipo familiar o de aparición esporádica) o a una disfunción secundaria a estados de disproteinemia, de hipervitaminosis D, de hipertiroidismo, etcétera.
Síndrome linfogranular mucocutáneo. Síndrome de una dermatitis confluente de la infancia. Se inicia como un cuadro febril agudo con poliadenopatías, al que se agrega una erupción maculoeritematosa que se caracteriza por ser confluente y edematizante, predominando en manos y pies y por la abundante descamación que sigue a la resolución de las lesiones. Fue observado por primera vez en Japón y su causa no se conoce.
Síndrome de Loffler. Síndrome pulmonar con manifestaciones alérgicas difusas. Se caracteriza por disnea asmatiforme con tos intensa y rebelde, leucocitosis leve o moderada con marcada eosinofilia y presencia de granulomas pulmonares múltiples que se manifiestan como infiltrados difusos, fugaces, en las radiografías, del tórax. La etiología del cuadro aún no se conoce con precisión.
Síndrome de Louis-Bar. Síndrome de la ataxia-telangiectasia. Se caracteriza por ataxia cerebelosa progresiva, telangiectasias oculocutáneas, infecciones frecuentes de los senos paranasales y los pulmones, y movimientos anormales de los ojos. Suele existir una deficiencia inmunitaria. Es una afección hereditaria que se trasmite como rasgo autosómico recesivo.
Síndrome de Lown-Ganong-Levine. Síndrome electrocardiográfico y clínico de excitación auriculoventricular anómala. Las manifestaciones clínicas, son las mismas que las del síndrome de Wolff-Parkinson-White, pero a diferencia que éste, la única anormalidad electrocardiográfica es un acortamiento del PR.
Síndrome de Lubarsch-Pick. Síndrome congénito producido por depósito de sustancia amiloide. Se caracteriza por el depósito de maternal, amiloide atípico en la piel y el tejido muscular, con esclerodermia y macroglosia. La causa es desconocida.
Síndrome de Lutembacher. Síndrome asociado a malformaciones cardíacas congénitas. Designa la combinación de comunicación interauricular (por persistencia del agujero oval), estenosis mitral e hipertrofia secundaria de la aurícula izquierda.
Síndrome de Lyell. Síndrome de la necrólisis epidérmica tóxica. Consiste en una dermatitis exfoliativa severa, precedida por eritema y grandes ampollas. Suele ser de causa infecciosa (estafilococos) en la infancia o tóxica (por drogas) en el adulto.
Síndrome del lleno alveolar. Síndrome radiológico de la ocupación de espacios alveolares. Se caracteriza por la presencia de imágenes nodulares borrosas medianamente densas, de límites difusos, que tienden a confluir en napa y que interesan un lóbulo o un segmento pulmonar, o que se distribuyen uniformemente en ambos campos pulmonares. Se lo observa en las bronconeumonías o en la fase inicial de las neumonías, en el cáncer alveolar, el edema agudo del pulmón, etcétera.
Síndrome de la macrogenitosomia precoz. ver Síndrome pineal.
Síndrome de Mallory-Weiss. Síndrome de la rotura esofágica por esfuerzo. Es un cuadro de hematemesis, a menudo masiva, que sobreviene en pacientes que han estado con vómitos repetidos y persistentes. Se produce a partir de laceraciones longitudinales en la vecindad de la unión esofagogástrica.
Síndrome de la mandíbula-guiño. ver Síndrome de Gunn.
Síndrome de Marcus Gunn. ver Síndrome de Gunn.
Síndrome de Marchiafava-Micheli. Síndrome de la hemoglobinuria paroxística nocturna. Se caracteriza por episodios de hemólisis paroxística con hemoglobinuria, habitualmente nocturnos, aumento de la hemoglobina plasmática y a veces leucopenia y trombocitopenia. Prevalece en adultos jóvenes y se debería a una mutación de las células madre de la médula ósea, con una exagerada sensibilidad al complemento.
Síndrome de Marfan. Síndrome producido por anomalías congénitas de los elementos fibrosos del tejido conjuntivo. Típicamente se manifiesta por: a) estatura alta, delgadez que a veces es extrema y tórax de conformación anormal; b) miembros gráciles y desproporcionadamente largos en relación con el tronco, lo cual es particularmente evidente en los segmentos más distales (aranodactilia); c) escafocefalia con paladar ojival y protrusión del acetábulo; d) anomalías oculares, que incluyen ectopia del cristalino, opacidades corneales y, en ocasiones, miopía y glaucoma; e) debilidad muscular, hiperlaxitud de las articulaciones y distintas deformaciones óseas, y f) marcada tendencia a desarrollar insuficiencia aórtica o aneurismas aórticos, como consecuencia de la debilidad congénita de la capa media de este vaso. Es un cuadro genético que se trasmite como un, rasgo autosómico dominante.
Síndrome de Marie-Bamberger. Síndrome de la osteoartropatía hipertrofiante néumica. Es secundario a neumopatías, crónicas y se caracteriza por hiperemia y edema del periostio, con neoformación de matriz ósea y posterior calcificación. Esto, produce un engrosamiento simétrico en muñecas y tobillos, metacarpianos y metatarsianos, y extremidades de los huesos largos suele haber “dedos en palillo de tambor”. Aún no se ha aclarado el mecanismo causal.
Síndrome de Maroteaux-Lamy. Síndrome de la mucopolisacaridosis VI. Sus características básicas son: a) facies de rasgos toscos, aunque menos acentuados que en el síndrome de Hurler, con talla, cuello y tronco cortos; b) hepatoesplenomegalia y tendencia al desarrollo de hernias, y c) ausencia de retardo mental, y desarrollo lento y tardío de opacidades corneales. El cuadro, que también tiene una forma leve, obedece a una deficiencia de aril sulfatasa B.
Síndrome de Mayer-Rokitansky-Kuster. Síndrome de amenorrea primaria de origen genético. Se caracteriza por ausencia o hipoplasia acentuada de la vagina, útero rudimentario asociado a veces a anomalías del aparato urinario y ocasionalmente a alteraciones esqueléticas. Tanto los caracteres sexuales secundarios como la función ovárica son normales, y si el útero rudimentario tiene endometrio, incluso puede haber hemorragia menstrual que al ser retenida produce dolores abdominales de aparición cíclica.
Síndrome de Meigs. Síndrome asociado a la presencia de tumores ováricos. Es la combinación de ascitis e hidrotórax con un tumor ovárico, por lo general un fibroma.
Síndrome de Melkersson-Rosenthal. Síndrome de la parálisis facial con tumefacción crónica de las partes blandas. Es un cuadro de parálisis facial recurrente asociado a edema facial crónico, persistente, que predomina en los labios y la lengua. La causa no ha sido aclarada.
Síndrome de Ménétrier. Síndrome de la obstrucción del conducto torácico. Edema duro, que afecta los miembros inferiores, la porción inferior del abdomen y el hemitórax y el brazo izquierdos, asociado a hidrotórax o quilotórax y derrame peritoneal. La causa suele ser tumoral.
Síndrome de Méniere. Síndrome del vértigo recurrente, acompañado de acúfenos y sordera. Suele iniciarse alrededor de los 50 años de edad, y se manifiesta por accesos agudos de vértigo, sordera que habitualmente es unilateral, y acúfenos en el oído afectado. La forma clásica obedece a una hemorragia laberíntica, mientras que en otros casos resulta de procesos degenerativos en el oído interno.
Síndrome meníngeo. Síndrome neurológico resultante de la irritación de las membranas meníngeas. El cuadro es típico y se manifiesta por: a) fotofobia y cefalea intensa, persistente y rebelde a toda medicación analgésica; b) vómitos fáciles, en chorro de tipo central, y constipación pertinaz; c) rigidez en extensión del tronco y el cuello, con hiperestesia cutánea e hiperreflexia muscular; d) trastornos de la conciencia, convulsiones, estado de coma, e) alteraciones cuantitativas y cualitativas del líquido cefalorraquídeo, (hipertensión, pleocitosis, presencia de sangre o de material purulento, etc.). Lo producen las hemorragias, infecciones y procesos irritativos.
Síndrome de la miastenia grave. Síndrome de debilidad muscular relacionado con alteraciones de la unión neuromuscular que impiden la trasmisión normal de los impulsos nerviosos. Sus rasgos básicos son: a) comienzo por lo general subagudo o insidioso; b) debilidad muscular que predomina en los músculos oculares (sin afectar las pupilas), en los músculos, faciales, laríngeos y faríngeos, y en el diafragma; c) fatiga muscular fácil y rápida al repetir movimientos o intentar mantener una postura, recuperación parcial después de un período de descanso y notable mejora en respuesta a la administración de agentes, colinérgicos (neostigmina), y d) no hay atrofia muscular, ni alteración de los reflejos, ni otras anomalías neurológicas, y el riesgo principal es la facilidad con que el individuo desarrolla episodios; de insuficiencia respiratoria. Existe una forma idiopática, la más frecuente, y otras que se asocian a tumores del timo, cuadros hipertiroideos y enfermedades del colágeno.
Síndrome mieloproliferativo. Denominación general de un grupo de afecciones caracterizadas por la proliferación incontrolada de los tipos celulares de la médula ósea. Define los signos y síntomas de cuadro procesos de evolución no aguda que se manifiestan por la proliferación predominante, más que aislada, de células de la línea granulocítica, megacariocítica, eritroide, y eventualmente, de fibroblastos. Son los siguientes: a) la leucemia granulocítica crónica, que cursa con gran esplenomegalia y cifras de granulocitos circulantes que a menudo llegan a los 200.000 por mm3; b) la trombocitemia hemorrágica esencial, donde predominan netamente los megacariocitos y la esplenomegalia es moderada; c) la policitemia vera, caracterizada por un aumento del número de células, circulantes de las tres series, pero fundamentalmente a expensas, de los eritrocitos y con hematócritos que por lo general superan el 60%, y d) la metaplasia mieloide agnógena o metaplasia mieloide-mielofibrosis, cuadro en el cual predominan los granulocitos, y megacariocitos pero con una fibrosis simultánea progresiva de la médula ósea, de modo que en las fases avanzadas cuando esta fibrosis, se ha completado, la proliferación celular tiene lugar en focos extramedulares (metaplasia) y explica la hipertrofia notable del hígado y el bazo que presentan estos pacientes. Estos cuatro cuadros tienen en común, además la posibilidad de que se produzcan transiciones de uno a otro y de que todos ellos, en la fase terminal adopten la forma de una leucemia granulocítica aguda o una leucemia indiferenciada.
Síndrome de Milroy-Nonne-Melge. Síndrome del linfedema crónico bilateral de los miembros inferiores. Se caracteriza por edema crónico y progresivo de las piernas, que comienza en las primeras épocas de la vida, asociado a veces a hemangiomas y linfangiomas. La afección es hereditaria, autosómica dominante; predomina en mujeres y obedecería a una hipoplasia congénita de los linfáticos.
Síndrome de Mikulicz. Síndrome de la hipertrofia de las glándulas salivales y lagrimales. Agrandamiento simétrico y habitualmente insidioso, e indoloro, de las glándulas salivales y lagrimales, que se acompaña de una supresión concomitante, parcial o completa, de la secreción de lágrimas y saliva. Existen formas idiopáticas y otras que aparecen en pacientes con sarcoidosis, linfomas, infecciones específicas, etc. La etiología es desconocida.
Síndrome de Milkman. Síndrome propio de ciertas formas de osteoporosis severa de la edad madura. Se lo observa predominantemente en mujeres y se caracteriza por a) descalcificación difusa con importantes alteraciones esqueléticas (cifosis acentuada, aplastamientos vertebrales, deformaciones óseas); b) dolor dorsal y en los miembros inferiores, a menudo de tipo radicular; c) debilidad y astenia crecientes, y d) anomalías características en las radiografías óseas (presencia de bandas trasversales radiolúcidas rodeadas por un borde opaco). La etiología del cuadro es desconocida.
Síndrome de Minkowski-Chauffard. Síndrome de la esferocitosis hereditaria. Se caracteriza por anemia de leve a moderada, esplenomegalia e ictericia por aumento de la bilirrubina no conjugada, debido a la presencia de esferocitos y de una mayor fragilidad eritrocitaria. Se trasmite como rasgo autosómico dominante.
Síndrome de Möbius. Síndrome neurológico de disgenesia de los núcleos de los nervios craneales. Se manifiesta por tipos y grados de parálisis o paresias que varían según el mayor o menor compromiso de los distintos nervios craneales inexpresividad de la cara, alteraciones de la motilidad ocular, dificultad para la succión, disfagia; disartria, etc. Ocasionalmente estos pacientes presentan sindactilia y atrofia de grupos musculares. La causa es desconocida.
Síndrome de von Monakow. Síndrome neurológico de la oclusión de la arteria coroideo anterior. Hemiplejía alterna acompañada de hemianopsia y hemianestesia o hemihipoestesia homónimas a raíz de lesiones que afectan el brazo posterior de la cápsula interna.
Síndrome de Mondor. Síndrome de las formas graves del aborto séptico. Se caracteriza por fiebre en picos, ictericia rápidamente creciente, hemoglobinuria hipotensión marcada, y oliguria que evoluciona casi siempre a la insuficiencia renal aguda. La facies suele ser típica: ictericia conjuntival palidez en las mejillas y cianosis peribucal.
Síndrome de Morgani-Stewart-Morel. Síndrome de la hiperostosis frontal interna. Consiste en engrosamiento de la tabla interna del hueso, frontal que afecta casi exclusivamente al sexo femenino, cerca de la menopausia, y se acompaña de obesidad, hipertricosis y distintas manifestaciones neuropsíquicas. Es de causa desconocida.
Síndrome de Morquio, Síndrome de la mucopolisacaridosis IV. Se distingue por: a) facies de rasgos moderadamente toscos, nariz ancha; boca amplia y alteraciones dentarias; b) talla y cuello cortos, cifosis acentuada y ensanchamiento anteroposterior del tórax; c) genu valgum con marcha de pato; d) hepatoesplenomegalia y eventualmente insuficiencia aórtica, y e) presencia de opacidades corneales, pero sin retardo mental. Se trasmite como un rasgo autosómico recesivo y obedece a una deficiencia de la sulfatasa del condroitinsulfato.
Síndrome de Moschcowitz. Síndrome purpúrico con trombosis diseminada. El cuadro es el de una anemia hemolítica grave de tipo extracorpuscular, asociada a una púrpura trombocitopénica y trastornos neurológicos múltiples (parálisis de nervios craneales, signos cerebelosos, vértigo y accesos convulsivos). La causa es desconocida.
Síndrome de las mucopolisacaridosis. Síndrome de la deficiencia congénita de enzimas específicas del metabolismo de los mucopolisacaridosis. Incluye un conjunto de cuadros clínicos que tienen en común: a) una deficiencia congénita de alguna de las enzimas esenciales para la degradación de tres tipos de mucopolisacáridos: el sulfato de heparán, el sulfato de dermatán y el sulfato de queratán; b )el carácter progresivo de la afección, cuyas manifestaciones se deben a la creciente acumulación de mucopolísacáridos en los distintos sistemas del organismo, y c) la existencia de ciertas, similitudes básicas en cuanto al fenotipo: facies de rasgos burdos, disostosis, múltiples, rigidez articular, hepatoesplenomegalia, excreción de mucopolisacáridos en la orina, etc. En cambio, se diferencian entre sí por la gravedad de las alteraciones esqueléticas, por la presencia o ausencia de ciertos componentes particulares (retardo mental, opacidades en la córnea), por la seriedad del pronóstico, y por la deficiencia enzimática propia de cada caso. A partir de combinaciones de estos elementos el grupo general de las mucopolisacaridosis ha sido subdividido en los siguientes tipos particulares: el tipo IH, o síndrome de Hurler; el tipo IS, o síndrome de Scheie (anteriormente: tipo V); el tipo II o síndrome de Hunter; el tipo III, o síndrome de Sanfilippo; el tipo IV o síndrome de Morquio; el tipo VI, o síndrome de Maroteaux-Lamy; el tipo VII, o síndrome de Sly; y el tipo VIII o síndrome de Di Ferrante.
Síndrome de la muerte súbita del lactante. Nombre con el que se designan los casos de muerte sorpresiva e inexplicable de un lactante. Típicamente, es el caso del niño que se duerme tranquilamente a la noche, y a la mañana siguiente es encontrado muerto en su cuna (cribcot-death, de los autores sajones), sin explicación alguna ni hallazgos que lo justifiquen, en los estudios necrópsicos. La causa es desconocida, aunque se han postulado distintos mecanismos que podrían ser responsables de un cuadro de este tipo: imposibilidad del niño, por inmadurez, de aumentar la ventilación en respuesta a obstrucciones respiratorias moderadas; intervención de un cuadro como el de la apnea del sueño del adulto; desencadenamiento de arritmias cardíacas y otros.
Síndrome de Munchhausen. Síndrome de fabulación en una personalidad histriónica. Es característico de individuos que solicitan ser tratados en centros hospitalarios alegando padecer una enfermedad aguda, sobre la cual ofrecen una historia coherente, a menudo dramática, pero falsa, y que reiteran esta conducta en las distintas situaciones de sus vidas.
Síndrome del muñón cístico. Síndrome posoperatorio de las colecistectomías. Incluye un conjunto de síntomas más o menos imprecisos, similares a los del síndrome poscolecistectomía salvo por la ausencia de ictericia, que se asocian a una dilatación del muñón residual y se atribuyen a fenómenos reflejos originados en esta zona.
Síndrome de Naegeli. Síndrome caracterizado por la asociación de hiperqueratosis anomalías pigmentarias y otras alteraciones cutáneas. Incluye hiperqueratosis palmoplantar, pigmentación reticulada de la piel, anhidrosis o hipohidrosis, y a veces alteraciones dentarias. Es de causa hereditaria y se trasmite como rasgo autosómico dominante.
Síndrome de Nelson. Síndrome consecutivo a ciertos tratamientos de la enfermedad de Cushing. Se lo observa en pacientes con enfermedad de Cushing a los que se efectuó una adrenalectomía bilateral con irradiación de la hipófisis. Consiste en el desarrollo de un tumor secretante de ACTH más agresivo que produce serias alteraciones del campo visual por invasión local.
Síndrome de las neoplasias endocrinas múltiples. Conjunto de manifestaciones que resultan de la hiperfunción simultánea de varios sistemas endocrinos diferentes. Designa una diversidad de cuadros clínicos asociados a la presencia de hiperplasias, o tumores endocrinos múltiples (pancreáticos; paratiroideos, hipofisarios, tiroideos y suprarrenales, además de neuromas) y cuyas manifestaciones son el resultado de la hiperfunción de las glándulas correspondientes. Si bien las variedades y combinaciones teóricas posibles son muchas, las formas concretas de presentación han permitido establecer tres grupos distintos dentro del síndrome de las neoplasias endocrinas múltiples: el tipo I o síndrome de Wermer en el cual se presentan tumores tiroideos, suprarrenales e hipofisarios, hiperplasia de las paratiroides y rumores no insulínicos de los islotes de Langerhans, y al cual se tiende a incorporar el síndrome de Zollinger-Ellison, anteriormente considerado como una entidad separada; el tipo II o síndrome de Sipple, caracterizado por la asociación de un carcinoma medular de la tiroides; con feocromocitomas e hiperplasia paratiroidea; y el tipo III o síndrome de los neuromas mucosos múltiples, combinación de un feocromocitoma y un carcinoma medular de la tiroides con numerosos neuromas mucosos y diferentes anomalías corporales.
Síndrome neuroanémico. ver Síndrome de Lichtheim.
Síndrome de los neuromas mucosos múltiples. Tipo III del síndrome de las neoplasias endocrinas múltiples. Se define por la asociación de tumores tiroideos y suprarrenales (carcinoma medular de la tiroides, feocromocitoma), por el desarrollo de neuromas múltiples en las mucosas (boca, conjuntiva, esófago, tubo digestivo) y por la presencia de malformaciones corporales (hábito similar al del síndrome de Marfan, cifoescoliosis, genu valgum, pies cavos, prognatismo de partes blandas, etc.).
Síndrome de la neuropatía retrobulbar. Síndrome de ceguera reversible de etiología desconocida Se manifiesta en adolescentes o adultos jóvenes por disminución brusca de la agudeza visual en un ojo, que evoluciona rápidamente a la ceguera, seguida del mismo cuadro, pocas semanas después, en el otro ojo. La pupila y la retina son normales y no se encuentran alteraciones neurológicas que justifiquen el trastorno. La mayoría de los pacientes se recuperan espontáneamente, sin que queden secuelas o con defectos leves o puntiformes en el campo visual.
Síndrome del nevo basocelular. ver Síndrome de Gorlin-Goltz.
Síndrome del nódulo sinusal enfermo. ver Síndrome de la enfermedad del nódulo sinusal.
 Síndrome de Noonan |
Síndrome de Noonan. Síndrome hereditario caracterizado por hipogonadismo malformaciones cardíacas y otras anomalías corporales. Sus rasgos básicos son: a) talla y cuello corto, con hipertelorismo, implantación baja del cabello y tórax en escudo; b) hipogonadismo y criptorquidia; c) estenosis pulmonar congénita y ocasionalmente, estenosis aórtica, y d) en algunos casos, retardo mental leve o moderado. No hay alteraciones en el cariotipo.
Síndrome de Nothnagel. Síndrome neurológico del tronco cerebral con trastornos cerebelosos y compromiso del III nervio craneano. Se caracteriza por parálisis unilateral o bilateral parcial de la motilidad ocular (III nervio craneano), asociada a ataxia cerebelosa (lesión del pedúnculo cerebeloso superior). Es causado, habitualmente, por tumores del techo del mesencéfalo.
Síndrome del núcleo ambiguo y el tracto espinotalámico. ver Síndrome de Avellis.
Síndrome del núcleo de Deiters. ver Síndrome de Bonnier.
Síndrome de la obstrucción arterial aguda. Síndrome de la interrupción brusca del flujo arterial a un sector del organismo. Se caracteriza por dolor, frialdad, palidez, anestesia, impotencia funcional y desaparición del pulso, periférico. Obedece a un espasmo o a la presencia de un obstáculo orgánico en una arteria (trombosis, embolia, compresión exógena).
Síndrome de la oftalmoplejía migrañosa. Síndrome oftalmológico transitorio asociado a accesos de migraña. Se presenta en adultos jóvenes bajo la forma de uno o más ataques unilaterales de parálisis, de los movimientos oculares (III y VI nervios craneanos) asociados a un cuadro de migraña, cuadro que por lo demás no se diferencia en nada de la forma clásica del ataque de migraña. Los pacientes curan espontáneamente y la causa se desconoce, aunque el síndrome ha sido atribuido al espasmo prolongado de alguna rama de la arteria oftálmica.
Síndrome de Ogilvie. Síndrome de la obstrucción aparente del colon. Se manifiesta por estados de contractura persistente de la musculatura del intestino grueso, sin causa orgánica que los justifiquen. Se lo atribuye a anomalías de la inervación simpática.
Síndrome de Oppenheim. Síndrome de las afecciones que evolucionan con flaccidez muscular en el paciente pediátrico. Denominación general, puramente clínica e independiente de la etiología, de todas las afecciones del recién nacido y de la infancia que cursan con disminución del tono de los músculos esqueléticos, y de las cuales el síndrome de Werdnig-Hoffinann es la entidad más conocida.
Síndrome de Ostrum-Frost. Síndrome caracterizado por malformaciones craneocervicales y de la cintura escapular. Es una tríada constituida por sinostosis congénita del cuello, invaginación occipital con protrusión de las vértebras cervicales en la fosa posterior (platibasia) y elevación congénita de la escápula (deformidad de Sprengel).
Síndrome de los ovarios poliquísticos. ver Síndrome de Stein-Leventhal
Síndrome de Pancoast. Síndrome neuromuscular de los tumores del vértice pulmonar. Se define como un cuadro de dolor reumático en el miembro superior, a veces con atrofia muscular focal, acompañado de miosis y parálisis del elevador del párpado superior. Es propio de las neoplasias cercanas al vértice pulmonar que invaden las raíces del plexo braquial.
Síndrome de la pancreatitis crónica. Síndrome de la inflamación crónica del páncreas. Se manifiesta por: a) episodios recurrentes de dolor abdominal de leve a moderadamente intenso y de duración variable (horas o días); b) esteatorrea y diabetes latente o manifiesta, de instalación insidiosa y evolución progresiva, y c) presencia de depósitos cálcicos en la totalidad del órgano, visibles en las radiografías. El cuadro predomina en individuos alcohólicos, obesos o con litiasis de las vías biliares, aunque también existen formas idiopáticas.
Síndrome de la pancreatitis hereditaria. Síndrome infantil de la pancreatitis. Se define por la aparición de ataques reiterados de dolor abdominal intenso, que duran de días a semanas, con aumento de los niveles plasmáticos de amilasa y lipasa, y por el desarrollo progresivo de las manifestaciones crónicas de la enfermedad: diabetes, esteatorrea y calcificación del páncreas. El cuadro es hereditario y se trasmite como un rasgo autosómico dominante con penetración incompleta.
Síndrome de Papp-Parkinson. Síndrome de la taquicardia paroxística repetitiva. Se caracteriza por la aparición de numerosísimas crisis breves de taquiarritmia auricular paroxística (taquicardia supraventricular, aleteo auricular, fibrilación auricular), separadas por intervalos también breves de ritmo sinusal con frecuentes extrasístoles auriculares. El cuadro es de causa desconocida, puede repetirse hasta 1000 veces por día, y lleva al individuo a un estado de virtual incapacidad.
Síndrome de la parálisis periódica familiar. Síndrome de la parálisis intermitente con hipopotasemia asociada. Se caracteriza por ataques recurrentes de parálisis fláccida de los músculos de los miembros, con arreflexia tendinosa y ausencia de respuesta muscular a la estimulación eléctrica. Los accesos suelen ocurrir a la noche, a menudo después de ingerir comidas abundantes en carbohidratos, y se acompañan, típicamente de una acentuada disminución de los niveles plasmáticos de potasio. Se trata de una afección familiar, de mecanismo no bien conocido, que permite al individuo desarrollar una vida normal en los períodos intercríticos.
Síndrome paramediano de Foix. ver Síndrome de Dejerine, def.2.
Síndrome paraneoplásico. Conjunto de manifestaciones asociadas a la presencia de un tumor maligno en órganos y sistemas no afectado directamente por el crecimiento neoplásico. Comprende distintos cuadros que a menudo preceden a la manifestación clínica del tumor, o que la acompañan, y que incluyen síndromes: a) endocrinos (de Cushing, ginecomastia, hjpertiroidismo, hipoglucemia, hiperpigmentación, hiperparatiroidismo, etc.) b) reumatiformes (polimiositis, osteoartropatía hipertrofiante, poliartritis, etc.); c) neurológicos (trastornos cerebelosos o cerebrales), d) hematológicos (trombosis, diseminadas) y otros. Ejemplos de esto son la asociación de hiperparatiroidismo con tumores renales, pancreáticos u ováricos; de ginecomastia o hiperpigmentación con carcinomas del pulmón; de hipertiroidismo con el coriocarcinoma o con el carcinoma embrionario del testículo; y de la acantosis nigricans con linfomas o adenocarcinomas.
Síndrome paratrigeminal de Raeder. Síndrome doloroso unilateral de la cara. Se caracteriza por crisis unilaterales de dolor facial intenso, localizado en la región maxilar y frontotemporal, y acompañado de hipoestesia y desigualdad pupilar. A diferencia de la neuralgia del trigémino, es producido por lesiones orgánicas (tumores; granulomas).
Síndrome de Parinaud, Síndrome neurológico, del tronco cerebral con compromiso de nervios craneanos. Se caracteriza por parálisis de la mirada hacia arriba, pupilas fijas y divergencia de los ojos. Lo produce la lesión de los tubérculos cuadrigéminos anteriores y del mecanismo supranuclear de coordinación de la mirada hacia arriba y la causa suele ser un pinealoma.
Síndrome parkinsoniano. Síndrome neurológico de la parálisis agitante. Es un cuadro, de comienzo insidioso y evolución progresiva cuyos rasgos básicos son: a) facies inexpresiva típica, seborreica y con salivación exagerada; b) rigidez muscular generalizada; c) temblor grueso involuntario, presente en reposo pero que desaparece durante el sueño y tiende a desaparecer con los movimientos; d) marcha también característica (marcha festinante), rápida, de pasos cortos y con el cuerpo inclinado hacia adelante. No hay alteraciones en el intelecto ni en los órganos de los sentidos. Es causado por lesión de los núcleos pigmentados del tronco cerebral (sustancia negra, locus coeruleus) y se lo observa como entidad pura en la enfermedad de Parkinson, y asociado a otras manifestaciones en cuadros de distinta etiología (parkinsonismo aterosclerótico, parkinsonismo posencefalítico, etc.).
Síndrome de Paterson-Kelly. ver Síndrome de Plummer-Vinson.
Síndrome peduncular de Claude. Síndrome neurológico del tronco cerebral con compromiso del III nervio craneal y alteraciones cerebelosas y de la articulación de la palabra. Se caracteriza por parálisis, de la motilidad ocular del lado de la lesión (III nervio craneano), acompañada de hemiasinergia en el lado opuesto (lesión del núcleo rojo) y de disartria. Es causado por aneurismas, trombosis o tumores del techo del rnesencéfalo.
Síndrome de Pellegrini-Stieda. Síndrome de disfunción de la articulación de la rodilla. Cuadro crónico de dolor, tumefacción y limitación de los movimientos de la rodilla, con calcificación del ligamento colateral interno de dicha articulación. Es una secuela alejada de los traumatismos o de los hematomas traumáticos.
Síndrome de Pende. Síndrome de hiperfunción global de la adenohipófisis. Se manifiesta por: a) aumento moderado de la talla; b) tendencia a la obesidad, preferentemente a nivel de las caderas y muslos; c) hiperplasia mamaria en la mujer y ginecomastia en el hombre; d) signos de craneopatía metabó1ica, y e) estrías cutáneas: similares a las de la enfermedad de Cushing. No hay hipertensión arterial y la causa del trastorno se desconoce.
Síndrome de Pendred. Síndrome hereditario caracterizado por sordera y alteraciones de la función tiroidea. Se manifiesta por sordera bilateral congénita y por el desarrollo posterior durante la primera infancia, de un bocio no hipotiroideo. El trastorno se observa en regiones en las que no existe bocio endémico y su causa es desconocida, aunque se lo atribuye a una incapacidad congénita para sintetizar normalmente las hormonas tiroideas.
Síndrome de Penfleld. Síndrome de hiperfunción neurovegetativa episódica asociada a una disritmia cerebral. Se caracteriza por accesos breves de inquietud, con fenómenos vasomotores en el territorio del simpático cervical, hipertensión arterial, lacrimación diaforesis, miosis o midriasis pupilar, taquicardia, bradipnea y en ocasiones pérdida del conocimiento. Fue atribuido por Penfield a descargas epilépticas originadas en el núcleo dorsal del tálamo.
Síndrome perisilviano posterior. ver Síndrome de la afasia sensorial
Síndrome de Peutz-Jeghers. Síndrome hereditario caracterizado por alteraciones cutáneas y tendencia al desarrollo de procesos malignos. Se manifiesta por la presencia de máculas pigmentadas en los labios, la mucosa bucal y los dedos, y por una poliposis intestinal que predomina en el intestino delgado. La afección se trasmite como un rasgo autosómico dominante y se ha demostrado una tendencia aumentada al desarrollo de adenocarcinomas gástricos, duodenales y del colon.
Síndrome de Pick. Síndrome de la pericarditis crónica constrictiva. Es un cuadro de hipertensión intrapericárdica crónica, consecutivo a una pericarditis reumática o tuberculosa, en el cual existe fibrosis hepática e ingurgitación yugular, pero sin latidos ni oscilaciones respiratorias. En la radioscopia no se observan los movimientos característicos de la actividad cardíaca ("corazón quieto").
 Síndrome de Pickwick |
Síndrome de Pickwick. Síndrome de hipoventilación pulmonar asociado a obesidad. Se caracteriza básicamente, por obesidad, somnolencia diurna marcada, policitemia y apetito exagerado, con aumento de la PC02 arterial y sin signos de enfermedad pulmonar orgánica subyacente; además, suele haber cianosis y respiración periódica nocturnas, y eventualmente, signos de insuficiencia cardíaca derecha. Las manifestaciones mejoran notablemente, cuando el paciente reduce su peso.
Síndrome de las piernas inquietas. Síndrome de una de las variedades del insomnio. Se caracteriza por: a) la necesidad virtualmente irreprimible del individuo de mover sus piernas cuando está inmóvil o sentado, y muy particularmente cuando se acuesta e intenta dormir; b) la explicación que proporciona sobre estos movimientos, que relaciona con un malestar profundo, intenso y sumamente desagradable que experimenta en los tobillos, y del cual intenta liberarse moviendo las piernas; c) la desaparición de la sensación después de haber dormido, y d) en todos los casos, la posibilidad de registrar, durante el sueño, movimientos rítmicos estereotipados, de dorsiflexión de los pies y de los dedos de los pies (movimientos periódicos del sueño), que son característicos de distintos tipos de insomnio. La causa es desconocida, si bien se atribuye a alteraciones neurofisiológicas reales, aunque todavía incompletamente conocidas.
Síndrome pineal. Síndrome por el que se manifiestan ciertos tumores del timo. Se caracteriza por crecimiento anormal temprano de los huesos largos, desarrollo, precoz de los genitales externos y de la función sexual, e hipertensión endocraneana.
Síndrome del piso orbitario. ver Síndrome de Dejean.
Síndrome de Plummer-Vinson. Síndrome de la anemia hipocrómica idiopática con aquilia. Se lo observa casi exclusivamente en mujeres y se manifiesta por: a) anemia hipocrómica que responde satisfactoriamente a la administración de cantidades elevadas de hierro; b) aquilia gástrica; c) atrofia de la mucosa de la boca, la faringe y el esófago superior, con disfagia y glositis; y característicamente, d) presencia de un pliegue en forma de media luna en la región cricofaríngea. La etiología del cuadro es desconocida.
Síndrome poscolecistectomia. Síndrome imprecisamente definido de ciertos pacientes sometidas a una colecistectomía. Conjunto de síntomas a menudo vagos y mal definidos que incluyen malestar o dolor difuso o de tipo cólico en el cuadrante superior derecho del abdomen, intolerancias alimentarias, sensación de plenitud, etc., y en ocasiones, ictericia. En un sentido estricto, la denominación debería aplicarse solamente a aquellos casos en que se demuestre que las manifestaciones son secuela de operaciones incompletas (litiasis residuales), de omisiones intraoperatorias (neoplasias que pasaron inadvertidas) o de complicaciones específicas (estenosis posquirúrgicas). Sin embargo, por costumbre más que por extensión, también se lo aplica a pacientes con molestias que sugieren un, trasfondo emotivo o funcional por el solo hecho de que hayan sido operados, y a otros que se quejan de los mismos síntomas que tenían antes de la intervención, lo cual induce a pensar que el diagnóstico fue incorrecto, la operación innecesaria, y que la causa real persiste (hernia hiatal, pancreatitis crónica, úlcera gástrica, etc.).
Síndrome poscomisurotomía. Síndrome posoperatorio alejado de la comisurotomía por estenosis mitral. Es un cuadro relativamente frecuente en este tipo de operaciones, que aparece de uno a tres meses después de la intervención y se manifiesta por fiebre, dolor retroesternal, artralgias difusas, a menudo migratorias, arritmias cardíacas; signos de poliserositis (pericarditis, pleuritis), etc. La causa no ha sido establecida con precisión.
Síndrome posflebítico. ver Síndrome postrombótico.
Síndrome posgastrectomía. ver Síndrome del vaciamiento rápido
Síndrome posinfarto de miocardio. ver Síndrome de Dressler.
Síndrome posirradiación. ver Síndrome por irradiación.
Síndrome posperfusión. Síndrome de la infección por citomegalovirus. Es un cuadro febril agudo con hepatomegalia, esplenomegalia y presencia de linfocitos atípicos, que se observó inicialmente en pacientes sometidos a intervenciones cardíacas con circulación extracorpórea. Posteriormente se demostró que el agente causal era un citomegalovirus y que las vías posibles de trasmisión eran muchas.
Síndrome postaquicárdico. Síndrome electrocardiográfico residual de las taquicardias ventriculares. Se lo observa después de los ataques prolongados y se caracteriza por la aparición de ondas T invertidas y ensanchadas que persisten así durante un tiempo variable (horas o días) para finalmente desaparecer. Es una manifestación del proceso de recuperación funcional y carece, por sí sola, de significado patológico.
Síndrome postrombótico. Síndrome crónico de las trombosis venosas de los miembros inferiores. Es un cuadro dermatológico que se observa en pacientes que han sufrido trombosis repetidas, y se caracteriza por una primera fase en la que predominan el edema y las hemorragias subcutáneas; una segunda fase con hiperpigmentación por hemosiderina, fibrosis subcutánea, atrofia de la piel y obstrucción linfática; y una tercera fase en la que aparecen úlceras: crónicas, recidivantes y de difícil curación.
Síndrome de Pourfour du Petit. Síndrome de la irritación del simpático cervical. Es el cuadro opuesto al del síndrome de Horner y se manifiesta por exoftalmía, midriasis y aumento de la hendidura palpebral. Es el resultado de un fenómeno irritativo, habitualmente por compresión o infiltración tumoral.
Síndrome premotor. Síndrome neurológico de las lesiones de la corteza cerebral premotora. Se manifiesta por hemiplejía espástica con hiperref1exia osteotendinosa, pérdida o disminución de la capacidad de realizar movimientos finos, trastornos vasomotores pasajeros, y actitudes del tipo de la aprehensión compulsiva. Es producido, casi siempre, por procesos tumorales expansivos.
Síndrome de Profichet. Síndrome caracterizado por calcificaciones subcutáneas y alteraciones musculares. Se manifiesta por la formación gradual de nódulos cálcicos bajo la piel, especialmente alrededor de las grandes articulaciones, seguida de atrofia muscular, ulceraciones cutáneas y trastornos neurológicos. Predomina en individuos jóvenes y la causa no ha sido establecida.
Síndrome protuberancial superior. ver Síndrome de Raymond-Cestan.
Síndrome de Putnam-Dana. ver Síndrome de Lichtheim.
Síndrome QT. Síndrome caracterizado por anomalías electrocardiográficas y episodios de taquiarritmia paroxística. Se aplica a aquellos casos en que un intervalo QT anormalmente prolongado, de carácter congénito y presente en el ritmo sinusal normal, se asocia a episodios paroxísticos de taquicardia ventricular polimorfa ("torsade de points"), en los cuales el tránsito desde el ritmo sinusal normal hacia la arritmia se caracteriza por la deformación cada vez más acentuada de los sucesivos complejos QRS, hasta que se establece el patrón característico de la taquicardia ventricular. Incluye los síndromes de Jervell y Lange-Nielsen, de trasmisión autosómica recesiva, acompañado de sordera nerviosa, y el de Romano-Ward, de herencia autosómica dominante y sin sordera nerviosa.
Síndrome quiasmático. Síndrome de las lesiones del quiasma óptico. Se manifiesta por cefalea, vértigos y vahídos, pérdida progresiva de la visión, a menudo no sistematizada y dependiendo de la mayor o menor medida en que vayan siendo afectados los distintos haces; y reducción del campo visual. La causa es tumoral.
Síndrome de Ramsay-Hunt. Síndrome de las afecciones virósicas del ganglio geniculado. Se manifiesta por una parálisis facial grave, unilateral, acompañada de otalgia y de una erupción vesiculosa, en ramillete, que afecta al conducto auditivo externo y el pabellón auricular. Obedece a la lesión del ganglio geniculado producida por el virus del herpes zoster.
Síndrome de Raymond-Cestan. Síndrome neurológico por lesión de la protuberancia. Se caracteriza, del lado opuesto al de la lesión, por hemiparesia, hemianestesia temblor estático, y asinergia de tipo cerebeloso, y por parálisis de los movimientos de lateralización de la mirada que predomina del lado de la lesión. Suele ser de causa vascular y lo producen las lesiones que afectan la porción alta de la protuberancia anular.
Síndrome de Raynaud. Síndrome caracterizado por accesos de vasoconstricción paroxística. Designa un cuadro de episodios de isquemia paroxística, fundamentalmente de los dedos de las manos, que se presentan, característicamente, como una reacción trifásica: primero palidez, luego cianosis y finalmente enrojecimiento a menudo con dolor y parestesias durante la primera fase. Desde el punto de vista clínico y para el mismo síndrome corresponde separar la enfermedad de Raynaud, trastorno funcional benigno y simétrico que predomina en mujeres jóvenes y que suele ser desencadenado por el frío o la emoción, del fenómeno de Raynaud secundario, que acompaña a otros procesos patológicos. Este último no tiene distinción de sexo y edad, puede ser asimétrico, a veces llega a provocar úlceras digitales, y se lo observa en ciertas colagenopatías; (esclerodermia), en la intoxicación por plomo y arsénico, durante la administración de distintos medicamentos (ergotamina metisergida, propranolol), en enfermedades hematológicas; (por crioaglutininas); en carcinomas ocultos, etc.
Síndrome de Reichmann. Síndrome de hipersecreción gástrica. Designa el estado caracterizado por hipersecreción de jugo gástrico ácido, cualquiera que sea su causa e independientemente de las eventuales complicaciones.
Síndrome de Reifenstein. Síndrome de seudohermafroditismo masculino asociado a hipogonadismo primario. Se caracteriza por eunucoidismo, ginecomastia e hipospadias, y por atrofia de los túbulos seminíferos con ausencia de espermatozoides. El trastorno obedece a anomalías en la síntesis o en la acción periférica de la testosterona, se manifiesta en la edad pospuberal, y es trasmitido como un rasgo recesivo ligado a X.
Síndrome de Reye. Síndrome paravirósico de la infancia con alteraciones hepáticas neurológicas y renales. Se presenta en niños previamente sanos que padecen infecciones de las vías respiratorias superiores, y se caracteriza por: a) signos de daño hepatocelular severo, con hipoglucemia,, acidosis, hiperamoniemia y aumento notable de las transaminasas séricas; b)encefalopatía grave con vómitos, edema cerebral y alteraciones rápidamente progresivas del sistema nervioso central; c) presencia de vacuolización extrema de las células hepáticas y de los túbulos renales, y d) elevada mortalidad, pero con recuperación compleja en los pacientes que sobreviven. Se lo observa como complicación de infecciones virósicas (por virus; de la influenza A, de la varicela, del grupo Coxsackie, y otros) cuyo curso inicial no difiere del habitual para cada tipo, de enfermedad, y casi siempre en pacientes que han sido tratados con aspirina. Por este motivo, se ha postulado la intervención de un mecanismo lesivo para las mitocondrias hepáticas en el cual la acción deletérea del salicilato y del agente infeccioso se vería potenciada por una deficiencia enzimática no detectada hasta ese momento, pero probablemente preexistente.
Síndrome de Riley-Day. Síndrome de la disautonomíafamiliar. Se caracteriza por inestabilidad autónoma (transpiración anormal, pérdida del control vasomotor, hipertensión lábil), trastornos del gusto, disminución de las sensaciones de dolor y temperatura, hiporref1exia, fiebre episódica, accesos, de vómitos y falta de lagrimación. La mayoría de los afectados no llega a la edad adulta. Se debería a una deficiencia enzimática aún no precisada.
Síndrome del robo subclavio. Síndrome de isquemia en el territorio vertebrobasilar, debido a estenosis -antes del nacimiento de la vertebral- de una de las arterias subclavias. Se caracteriza por la aparición de mareo, síncope u otras manifestaciones de insuficiencia vertebrobasilar, cuando el paciente hace ejercicio con el brazo del lado afectado. En estas circunstancias, en las cuales la subclavia no alcanza a aportar la sangre que el miembro en actividad necesita, se produce una inversión compensatoria del flujo en el sistema vertebral (del encéfalo pasa a la subclavia, más allá de la oclusión, y de allí al brazo) y aparecen los síntomas; es decir, la subclavia "roba" la sangre que debía ir al encéfalo.
Síndrome de Rochon-Duvigneau. Síndrome neurológico de la lesión unilateral de los nervios craneanos III, IV, V y VI. Suele ser un cuadro, insidioso y progresivo que lleva a la oftalmoplejía completa (nervios craneanos III, IV y VI), con anestesia de la córnea (rama oftálmica del V nervio craneano). Es causado por aneurismas, meningiomas y otros tumores que crecen en la vecindad de la hendidura esfenoidal
Síndrome de Roger. Síndrome de los tumores malignos del esófago. Designa el cuadro de sialorrea continua que acompaña a las neoplasias del esófago o de la región esofagofaríngea.
Síndrome de Romano-Ward. ver Síndrome QT
Síndrome de Rothmann-Makai. Síndrome de la inflamación circunscrita del tejido, graso subcutáneo. Designa un cuadro benigno de paniculitis circunscrita, que se manifiesta por necrosis de las células grasas y formación de granulomas adiposos que se trasforman en estructuras quísticas. La causa se desconoce.
Síndrome de Rothmund-Thomson. Síndrome esclerodérmico familiar con telangiectasias cutáneas y alteraciones orgánicas múltiples. Se caracteriza por el desarrollo de: a) una esclerosis progresiva en bandas, simétrica, que predomina en la cara y en los dedos y lleva rápidamente a la rigidez; b) telangiectasias, cutáneas con alteraciones pigmentarias asociadas; c) cataratas y signos generalizados de envejecimiento prematuro, d) ulceraciones tróficas y e) hipogonadismo y otras anomalías. Se trasmite como un rasgo autosómico recesivo.
Síndrome de la rotura inminente de útero. ver Síndrome de Bandl-Frommel
Síndrome de Roussy-Lévy. Síndrome neuromuscular con manifestaciones centrales y periféricas. Designa la combinación de una atrofia muscular progresiva, de origen neuropático y que predomina en la región peronea, con ataxia cerebelosa y escoliosis de la columna dorsal y lumbar.
Síndrome de Sanfilippo. Síndrome de la mucopolisacaridosis III. Se caracteriza, dentro del grupo de las: mucopolisacaridosis por el notable predominio de las alteraciones neurológicas en comparación con las somáticas, que son relativamente leves. Sus rasgos básicos son: a) facies de rasgos toscos, cejas hirsutas, boca entreabierta y anomalías dentarias múltiples; b) rigidez articular moderada con alteraciones esqueléticas mínimas (cuello y dedos cortos); c) hepatoesplenomegalia, y d) sordera, retardo mental severo y ausencia de opacidades corneales. Se trasmite como un rasgo autosómico dominante, obedece a deficiencia de distintas; enzimas (heparansulfatasa, N-acetil-D-glucosaminidasa, etc.) y se han descrito cuatro variantes distintas: (tipos A, B, C y D).
Síndrome de Sayre-Kearns. Síndrome caracterizado por la asociación de miopatía ocular, miopatía miocárdica y retinitis pigmentaria. Se define por el desarrollo de una oftalmoplejía bilateral progresiva que respeta la pupila y los músculos de la acomodación, acompañada de retinitis pigmentaria y de una miopatía cardíaca concomitante, y por la presencia de una cantidad llamativa de mitocondrias en los tejidos afectados. La causa es desconocida.
Síndrome de Scheie. Síndrome de la mucopolisacaridosis IS. Se caracteriza por: a) rasgos faciales toscos, con boca grande, narinas amplias y cejas hirsutas; b) rigidez articular que aparece alrededor de los 10 años, con trastornos esqueléticos menores e insuficiencia aórtica en épocas posteriores de la vida; c) presencia de opacidades corneales, y d) inteligencia normal. Se trasmite como un rasgo autosómico recesivo y obedece, al igual que el síndrome de Hurler, a una deficiencia de alfa-L-iduronidasa, pero a diferencia de éste, los individuos afectados pueden tener una larga sobrevida.
Síndrome de Schmidt. Síndrome neurológico de la lesión unilateral de los núcleos ambiguo y accesorio del X nervio craneano. Se define por la presencia de parálisis de la cuerda vocal, del velo del paladar, y de los músculos trapecio y esternocleidomastoideo, del mismo lado de la lesión. La causa suele ser vascular o tumoral
Síndrome de Schwachmann. Síndrome de hipoplasia congénita del páncreas con anomalías hematológicas. La manifestación dominante es un cuadro de diarrea con esteatorrea, similar al que se observa en la fibrosis quística del páncreas, acompañado de alteraciones hematológicas cuya causa radica en la médula ósea (neutropenia, y con menos frecuencia, anemia y trombocitopenia), y ensombrecido por infecciones intercurrentes que a menudo son severas. A diferencia de la fibrosis quística del páncreas la concentración de solutos en el sudor es normal y no existen trastornos respiratorios. Se trasmite como un rasgo autosómico dominante.
Síndrome de Seabright-Bantam. Síndrome del seudohipoparatiroidismo. Sus rasgos básicos son: a) enanismo, obesidad y retardo mental de grado variable; b) facies redonda, y cuello, tronco, extremidades y dedos cortos; c) presencia de calcificaciones diseminadas; d) hipocalcemia, crisis de tetania y accesos convulsivos; y e) hiperplasia paratiroidea con niveles elevados de parathormona circulante. El cuadro es familiar de causa desconocida, y se debe a una resistencia primaria de los órganos efectores (huesos y riñones) a la acción, de la hormona paratiroidea endógena.
Síndrome de Selye. ver Síndrome general de adaptación.
Síndrome de Senear-Usher. Síndrome del pénfigo eritematoso. Consiste en lesiones crónicas de aspecto seborreico, que predominan en cara y tronco y habitualmente no afectan las mucosas. La evolución es más larga que la del pénfigo vulgar.
Síndrome del seno carotídeo. Síndrome de la estimulación del seno carotídeo. Es un cuadro brusco de vértigo lipotimia o síncope, a veces con convulsiones, desencadenado por la estimulación mecánica accidental de un seno carotídeo (compresión digital, rotación brusca y exagerada del cuello). La hipotensión arterial se acompaña de bradicardia, característica de los episodios vasovagales.
Síndrome del seno cavernoso. ver Síndrome de Foix.
Síndrome seudobulbar. Síndrome neurológico asociado a lesiones difusas del sistema nervioso central. Se define por la presencia de trastornos neurológicos y psíquicos múltiples, y en particular de la mímica, la deglución y la fonación, producidos por lesión de los núcleos de la base, de las regiones faciales de la corteza cerebral o de los haces geniculados. En general no se presenta como un síndrome puro, sino como parte de un cuadro de deterioro, cerebral global, producido por lesiones vasculares difusas, o por focos múltiples de reblandecimiento.
Síndrome de seudoclaudicación intermitente. Síndrome de claudicación de la marcha de causa neurológica. Es un cuadro fácilmente confundible con el de la claudicación intermitente de origen vascular (dolor que aparece durante la marcha y que se alivia con el reposo), pero del cual se diferencia por: a) la relación entre la distancia recorrida y la aparición del dolor, que no es constante como en el caso de la claudicación isquémica, sino sumamente variable, y entre el comienzo del reposo y la desaparición del dolor que no ocurre con rapidez, sino lentamente y en un tiempo también variable; b) la distribución del dolor, que predomina en el muslo y en la región glútea y no en los tobillos; c) la coexistencia de un dolor lumbar más o menos permanente, que a menudo persiste en el decúbito, y d) la normalidad de los pulsos periféricos. El cuadro obedece a una compresión habitualmente subaguda, lentamente progresiva, de raíces nerviosas, y es causado por hernias de disco, deformaciones artrósicas, o tumores de la médula espinal.
Síndrome del seudocoma. Síndrome neurológico caracterizado por inmovilidad y falta de respuesta a los estímulos similares a los del coma, pero con conservación de la conciencia y producido por una interrupción virtualmente completa de las vías eferentes de la expresión corporal. Sus rasgos básicos son: a) es consecuencia directa de un accidente vascular encefálico; b) ni espontáneamente, ni en respuesta a estímulos, se producen movimientos de los miembros del tronco, de la cara o de la faringe, ni se emiten sonidos, tal como ocurre en el coma, pero con la particularidad de que persisten movimientos de pestañeo, y c) el individuo está consciente, como lo evidencian los movimientos de pestañeo, que realiza en respuesta directa a preguntas, del examinador. El síndrome es producido por trombosis de la arteria basilar en la región ventral de la protuberancia, que interrumpe la totalidad de las vías corticoespirales y corticobulbares descendentes, pero sin afectar el sistema reticular de activación (conciencia) ni las vías de la motilidad vertical del ojo (pestañeo).
Síndrome del seudohipoparatiroidismo. ver Síndrome de Seabright-Bantam
Síndrome seudomiasténico. ver Síndrome de Eaton-Lambert
Síndrome del seudotumor cerebral. Síndrome caracterizado por manifestaciones neurológicas equiparables a las de un tumor cerebral, pero sin patología orgánica detectable. Predomina en mujeres jóvenes y sus rasgos son los siguientes: a) cefalea moderada, lentamente progresiva, ocasionalmente acompañada de vértigos o de diplopía leve, en pacientes con excelente estado general; b) edema bilateral de papila asociado a estrechamiento del campo visual y agrandamiento del punto ciego, en ambos casos de grado leve; c) hipertensión del líquido, cefalorraquídeo, y d) tamaño normal o levemente disminuido de los ventrículos cerebrales, normalidad de los estudios tomo gráficos y arteriográficos, y ausencia de elementos atípicos en los análisis del líquido cefalorraquídeo. El cuadro es reversible, responde al tratamiento con corticosteroides, y su causa se desconoce.
Síndrome del seudoxantoma elástico. ver Síndrome de Groenblad-Strandberg.
Síndrome de Sézary. Síndrome de la fase leucémica de la micosis fungoide. Se lo observa en las etapas avanzadas de la micosis fungoide (linfoma cutáneo maligno), cuando ya se encuentran, células neoplásicas en la sangre circulante, y se define por la presencia de hiperpigmentación, placas dismórficas y engrosamiento difuso de la piel, y fundamentalmente, de una eritrodermia generalizada. En las fases terminales existe hepatoesplenomegalia junto con otras manifestaciones de invasión visceral.
Síndrome de Shy-Dragger. Síndrome de hipotensión ortostática con otras manifestaciones neurológicas. El cuadro, es el del síndrome de hipotensión ortostática idiopática al que se le agregan las manifestaciones propias de las lesiones cerebrales difusas. Se lo observa cuando predomina la degeneración de las vías extrapiramidales, de los ganglios basales y del núcleo dorsal del vago.
Síndrome de Sickert-Millikan. Síndrome de la oclusión intermitente de la arteria basilar. Se manifiesta por episodios recurrentes, transitorios y más o menos fugaces, de vértigo, pérdida de la visión, diplopia, disartria, disfagia y en ocasiones, hemiparesia.
Síndrome de Silvestrini-Corda. Síndrome hiperestrogénico de las hepatopatías crónicas avanzadas. Designa al conjunto de signos de feminización que se observa en la cirrosis, hepática avanzada, y que resultan de la incapacidad del hígado de inactivar los estrógenos circulantes: ginecomastia, hábito eunucoide, pérdida del vello pubiano y axilar, disminución de la libido, esterilidad y atrofia testicular.
Síndrome silviano frontal superior. ver Síndrome de la afasia motriz
 Síndrome de Stein-Leventhal. Obsérvese la hipoplasia mamaria, tendencia a la obesidad y virilización en un paciente de 16 años (Surós, Semiología Médica, 1968) |
Síndrome de Sipple. Tipo II del síndrome de las neoplasias endocrinas múltiples. Designa la presencia de un feocromocitoma (que a menudo es bilateral y múltiple), de un carcinoma medular de la tiroides, y de una hiperplasia paratiroidea. También pueden existir otros tumores: meningiomas, glioblastomas, etc. Los síntomas, dominantes suelen ser los del hiperparatiroidismo, y con menos frecuencia los del feocromocitoma. El trastorno es hereditario y se trasmite como un rasgo autosómico dominante.
Síndrome de la siringomielia. Síndrome neurológico de la gliomatosis de la sustancia gris de la médula espinal. La siringomielia es una proliferación de la neuroglia periependimaria (gliomatosis) que destruye la sustancia gris medular y afecta principalmente a los segmentos cervicales y lumbares. Se caracteriza, básicamente, por trastornos sensitivos (disociación siringomiélica pérdida de la sensibilidad térmica y al dolor, con conservación de la sensibilidad táctil y profunda), manifestaciones motoras (parálisis espástica) y alteraciones tróficas (artropatía siringomiélica, atrofias, de tipo Aran-Duchenne, etc.) La causa es desconocida.
Síndrome de Sjögren. Síndrome caracterizado por la tríada de artritis, xeroftalmía y xerostomía. Es un cuadro de sequedad ocular (xeroftalmía, queratoconjuntivitis seca) y sequedad bucal (xerostomía) que se presenta en individuos con artritis reumatoidea. Tiene un curso crónico y se lo atribuye a mecanismos de autoinmunidad.
Síndrome de Sluder. Síndrome de la neuralgia del ganglio esfenopalatino. Se caracteriza por dolor unilateral sordo y continuo en la región del maxilar superior, que se propaga al interior del cuello y el hombro, y se acompaña de obstrucción nasal, rinorrea y otalgia.
Síndrome de Sly. Síndrome de la mucopolisacaridosis VII. Es una forma parecida a un síndrome de Hurler leve y se caracteriza por: a) estatura corta con tórax en quilla; b) dismorfia facial con facies aplanada de rasgos toscos, nariz deprimida y surco subnasal alto; c) retardo mental moderado y d) inclusiones llamativas en los granulocitos. Es ocasionado por una deficiencia de beta-glucuronidasa y se transmite como un rasgo autosómico recesivo.
Síndrome de Spiller. Síndrome neurológico de la compresión crónica de la médula espinal. Es una secuela de la paquimeningitis de resolución incompleta (cicatrices remanentes) y se manifiesta por dolor y otros trastornos sensitivos, debilidad muscular, parálisis ascendente y signos de mielitis trasversa.
Síndrome de Steele-Rlchardson-Olszewski. Síndrome neurológico de la parálisis supranuclear progresiva. El comienzo es insidioso y se caracteriza por alteraciones del equilibrio, de la marcha y caídas inesperadas, rigidez que afecta principalmente al cuello y en menor medida a los músculos del tronco, dificultad para dirigir la mirada hacia abajo, y debilitamiento del volumen de la voz. En la etapa final hay rigidez extrema, oftalmoplejía completa, anartria e incapacidad total.
Síndrome de Stein-Leventhal. Síndrome de los ovarios poliquísticos. Se lo observa en mujeres jóvenes y se define por la asociación de ovarios poliquísticos, oligomenorrea y esterilidad, que se acompañan de grados variables de obesidad, hirsutismo e hipoplasia mamaria. Se atribuye a alteraciones en la síntesis de estrona.
Síndrome de Stevens-Johnson. Síndrome de una forma grave del eritema multiforme. Se caracteriza por el desarrollo de lesiones ulcerativas localizadas en las mucosas oronasal y anogenital, los ojos y vísceras, con cefalea, artralgia, fiebre y postración. Puede ser fatal. Existen casos idiopáticos y otros asociados a enfermedades virósicas o ingestión de distintas drogas.
Síndrome de Still. Síndrome de una de las formas de la artritis reumatoidea. Se refiere a un cuadro de poliartritis crónica acompañado de adenopatías generalizadas, episodios febriles recurrentes con rash cutáneo, y esplenomegalia; puede haber pericarditis, pleuritis y neumonitis. Existe anemia, leucocitosis y pruebas negativas para el factor reumatoideo y anticuerpos antinucleares. Las manifestaciones comienzan en la infancia o la adolescencia y la causa no se conoce con precisión.
Síndrome de Stokes-Adams. Síndrome asociado al bloqueo auriculoventricular. Se caracteriza por pérdida repentina de la conciencia, con convulsiones o sin ellas, debido a la falla del marcapaso inferior en el bloqueo AV completo.
Síndrome de Strachan. Síndrome caracterizado por trastornos neurológicos y alteraciones cutaneomucosas de origen nutricional. Define un cuadro de desarrollo progresivo de trastornos oculares (disminución de la capacidad visual, ceguera), signos de neuropatía periférica (parestesias, ataxia, pérdida de la sensibilidad superficial y profunda) y lesiones cutáneas y mucosas (glositis, estomatitis, etc.), que se observa en poblaciones humanas mal nutridas o sometidas a condiciones forzadas de restricción alimentaria (p.ej., en ciudades sitiadas).
Síndrome de Sturge-Weber-Dimitri. Síndrome de la angiomatosis encefalotrigeminal. Se define como la asociación de un angioma facial plano, de tipo capilar y ubicado en el territorio de distribución del trigémino, con un angioma meníngeo venoso, que se localiza con más frecuencia en la región parietooccipital. Clínicamente se manifiesta por crisis epilépticas sumamente frecuentes, a veces con hemiparesia y hemianopsia, y por retardo mental, mientras que las radiografías de cráneo muestran la presencia de calcificaciones lineales características.
Síndrome de Sudeck. Síndrome de la osteoporosis postraumática circunscrita. Designa los focos de osteoporosis localizada, acompañados de dolor y atrofia de los tejidos blandos, que se desarrollan a veces como consecuencia de pequeños traumatismos repetidos (p.ej., en los dedos de los pies), y más a menudo alrededor de las zonas de fractura durante el periodo de inmovilización.
Síndrome de Sweet. Síndrome de la dermatosis neutrofilia febril aguda. Se caracteriza por una erupción no pruriginosa de nódulos o placas rojos, dolorosos, en parte confluentes, que se inicia en brazos y suele afectar a mujeres; existe fiebre elevada y neutrofilia. La causa se desconoce.
Síndrome tabético. Síndrome neurológico de la sífilis avanzada. La tabes dorsal es una meningorradiculitis posterior que conduce a una rápida esclerosis de los cordones posteriores de la médula espinal. Sus manifestaciones básicas son: a) ataxia con marcha taloneante e hipotonía muscular; b) disociación de la sensibilidad de tipo tabético (pérdida de la sensibilidad profunda y táctil, con conservación de la sensibilidad al dolor y el calor); c) alteraciones tróficas (mal perforante plantar, artropatías tabéticas), y d) serología positiva para sífilis.
Síndrome de Takayasu. Síndrome de la oclusión progresiva de las ramas del cayado aórtico. Se caracteriza por una pérdida del pulso en los dos brazos y en las carótidas, junto con síntomas ocasionados por la isquemia cerebral (síncope, hemiplejía transitoria, etc.), ocular (ceguera transitoria, atrofia de retina, etc.), de la cara (atrofia muscular) y brazos (claudicación). Obedece a inflamación y obliteración progresiva del tronco braquiocefálico y de las arterias subclavia y carótida primitiva izquierdas, por encima de su origen en el cayado aórtico. Se supone una etiología autoinmune.
 Síndrome de Still |
Síndrome de Tapia. Síndrome neurológico de la lesión de dos núcleos de nervios craneales. Se manifiesta por parálisis unilateral de la laringe (núcleo ambiguo) y de la lengua (núcleo del hipogloso), sin parálisis del velo del paladar.
Síndrome de la taquicardia paroxística repetitiva. ver Síndrome de Papp-Parkinson.
Síndrome TAR. ver Síndrome trombocitopénico con ausencia del radio
Síndrome talámico. Síndrome de las lesiones del tálamo óptico. Según qué regiones estén predominantemente afectadas, el síndrome incluye combinaciones variables de algunas de las siguientes manifestaciones que se observan en el lado del cuerpo, opuesto al de la lesión: a) hemianestesia profunda total con hemianestesia superficial menos acentuada; b) dolor desagradable e intenso (dolor talámico) en el lado afectado, que aparece después del periodo agudo y en los casos en que resultó afectado el núcleo posteroinferior; c) alteraciones motoras, incluyendo hemiparesia transitoria, reducción de la motilidad voluntaria y aparición de movimientos anormales (temblor y ataxia, sincinesias de imitación movimientos coreicos y atetósicos); d) hemianopsia homónima, en las lesiones posteriores, y e) manifestaciones neuropsíquicas, con marcada reducción del lenguaje, perseveración verbal y de las ideas, etcétera.
Síndrome de Taussig-Bing. Síndrome de un cuadro de cardiopatía congénita con malformaciones complejas. Se define por: a) una tríada anatómica constituida por trasposición de los grandes vasos, comunicación interventricular y cabalgamiento de la arteria pulmonar, que producen una hipertrofia secundaria del ventrículo derecho; b) una saturación de oxígeno, que es mayor en la sangre de la arteria pulmonar que en la sangre de la aorta, y c) sus manifestaciones clínicas: hipertensión pulmonar, cianosis permanente, retardo, en el crecimiento, dedos en palillos de tambor, etcétera.
Síndrome de Thieberge-Weissenbach. Síndrome de la esclerodermia con calcificaciones difusas. Se define por la asociación de una esclerodermia generalizada con una calcinosis difusa, que se atribuye a una disfunción paratiroidea concomitante.
Síndrome de Thomson. ver Síndrome de Rothmund-Thomson.
Síndrome tibial anterior. Síndrome de la necrosis de los músculos tibiales. Se manifiesta por tumefacción rápida, tensión creciente, dolor y necrosis isquémica de la musculatura tibial anterior. La piel se ingurgita y se pone brillante, eritematosa y lustrosa a medida que progresa la necrosis. La causa es desconocida, aunque se supone relacionada con un exceso de ejercicio.
Síndrome de Tietze. Síndrome doloroso localizado en las articulaciones condrocostales. Predomina netamente en mujeres entre 35 y 50 años, y se caracteriza por dolor localizado en una de las articulaciones condroesternales, y con menos frecuencia, en una articulación esternoclavicular
Síndrome de Tolosa-Hunt. Síndrome doloroso unilateral de la cara. Se caracteriza por dolor facial unilateral, que predomina netamente en la región orbitaria (primera rama del trigémino), acompañado de oftalmoplejía y desigualdad de las pupilas. Es causado por procesos inflamatorios o tumorales situados en la vecindad de la hendidura orbitaria superior o del seno cavernoso.
 Síndrome de Touraine-Solente-Golé. Obsérvese el marcado engrosamiento de la piel y la hiperplasia de los tejidos blandos de los dedos con uñas curvadas (Modero Medicine, 1973) |
Síndrome de Toni-Fanconi. ver Síndrome de Fanconi.
Síndrome de Touraine-Solente-Golé. Síndrome de la paquidermoperiostosis. Consiste en un engrosamiento marcado de la piel de la cara, frente y cuero cabelludo, con formación de gruesos pliegues, neoformación perióstica y desarrollo de dedos en palillo de tambor. Se hereda como rasgo autosómico dominante y suele aparecer alrededor de la pubertad.
Síndrome de Treacher-Collins. Síndrome de un cuadro hereditario con alteraciones predominantemente maxilofaciales. Sus rasgos básicos son: a) hipoplasia bilateral de los huesos malares y del maxilar inferior; b) malformaciones palpebrales, que incluyen la presencia de un coloboma en el tercio externo del párpado inferior, y c) anomalías del pabellón auricular, a veces del oído medio y el interno, y eventualmente sordera. Se trata de un trastorno familiar en el cual las malformaciones se producen durante la vida intrauterina.
Síndrome tricorrinofalángico. Síndrome caracterizado por anomalías esqueléticas, del cabello y la nariz. Se caracteriza por braquidactilia y desviación axial de los dedos, escoliosis o lordosis, estatura baja, cabello escaso y delgado, y nariz piriforme con surco subnasal largo. La causa se desconoce y es compatible con una vida por lo demás normal.
Síndrome de triple X. Síndrome hereditario de individuos con retardo mental y amenorrea. Se presenta en mujeres y se define como la asociación de debilidad mental de grado variable, amenorrea secundaria, y anomalías corporales poco características. Las personas afectadas presentan una trisomía del cromosoma sexual X.
Síndrome de trisomía 21. ver Síndrome de Down.
Síndrome trombocitopénico con ausencia del radio. Síndrome de un cuadro combinado de malformaciones cardíacas y óseas, y alteraciones hematológicas. Se define por la presencia de una tetralogía de Fallot o de defectos en el tabique interauricular, acompañados de trombocitopenia y aplasia radial Es una afección hereditaria.
Síndrome de la trombosis capsular. Síndrome neurológico por lesión unilateral de la cápsula interna. Es el síndrome clásico, completo de la hemiplejía alterna, en la cual existe parálisis de la mitad del cuerpo y de la cara del lado opuesto al de la lesión cerebral. Lo produce la oclusión de las ramas perforantes de la arteria cerebral media.
Síndrome de Trousseau. Síndrome trombótico asociado, a la presencia de un proceso neoplásico. Aparición de trombosis venosas espontáneas, habitualmente repetidas, sin causa aparente y predominantemente en los miembros inferiores asociadas a la presencia de un carcinoma oculto. Se lo considera como un síndrome paraneoplásico.
Síndrome de los tubérculos cuadrigéminos. Síndrome neurológico de la lesión unilateral de los tubérculos cuadrigéminos. Sus manifestaciones son básicamente auditivas y oculares hipoacusia de tipo central acompañada de parálisis de los músculos dependientes del III nervio craneano, parálisis de la mirada vertical hacia arriba, paresia de la convergencia, nistagmo, y anisocoria con signo de Argyll-Robenson. La causa suele ser tumoral.
Síndrome del túnel carpiano. Síndrome neurológico periférico por compresión del nervio mediano. Se caracteriza por dolor y hormigueos o parestesias quemantes en la mano y los dedos, que puede producir degeneración de las fibras nerviosas y llevar a la atrofia muscular. Resulta de la compresión del nervio, mediano en el canal del carpo y a menudo es bilateral.
Síndrome del túnel tarsiano. Síndrome neurológico periférico por compresión del nervio tibial posterior. Obedece a la compresión de este nervio en el canal del tarso, o de sus ramas plantares, y se manifiesta por dolor, entumecimiento y parestesias en la planta del pie.
Síndrome de Turner. Síndrome caracterizado por agenesia gonadal y anomalías múltiples de causa genética (monosomía del cromosoma X, tipo XO). Sus rasgos son: a) talla reducida y cuello corto; b) linfedema de los miembros superiores e inferiores; c) agenesia gonadal con infantilismo sexual, genitales externos femeninos, hipoplasia mamaria y amenorrea primaria (en la mujer), y d) disminución de los estrógenos sanguíneos con aumento de las gonadotrofinas en la orina. El estudio de la cromatina sexual revela que algunos de estos pacientes son genéticamente "masculinos" y otros genéticamente "femeninos", más allá de que fenotípicamente no existan diferencias entre unos y otros.
Síndrome de Turner masculino. ver Síndrome de Noonan
Síndrome de Uehlinger. Síndrome caracterizado por la asociación de hiperostosis generalizada con engrosamiento localizada de la piel. El cuadro se define por el desarrollo de una osteoesc1erosis difusa que afecta a las epífisis, metáfisis, y diáfisis, y que se manifiesta por depósito subperióstico de cantidades anormales de hueso esponjoso por dolor tumefacción y rigidez de las articulaciones y por marcado engrosamiento de la piel de la parte inferior del brazo, y el antebrazo. Se inicia en la pubertad o adolescencia y la causa es desconocida.
Síndrome de la uña-rótula. Síndrome hereditario caracterizado por anomalías óseas y de las uñas y enfermedad renal. Sus rasgos son: a) codos en forma de cuña, rótulas rudimentarias y configuración en asta de los huesos ilíacos; b) uñas deformadas con hendiduras múltiples y c) signos de glomerulonefritis crónica moderada, por lo general de evolución benigna. El cuadro es de una osteosdisplasia, de causa desconocida, y se trasmite como un rasgo autosómico dominante.
Síndrome de Urbach-Wiethe. Síndrome de la proteinosis lipoidica. El cuadro obedece al depósito extracelular de material hialino en la piel y en la mucosa de la faringe y la laringe, y se manifiesta por: a) disfonía o afonía, macroglosia y anomalías dentarias; b) desarrollo progresivo de pápulas cutáneas de distribución generalizada, de color blanco amarillento y formadas por material intensamente positivo en la reacción del PAS, y c) presencia de calcificaciones simétricas en el área de la silla turca. Es una enfermedad granulomatosa por almacenamiento de lípidos que habitualmente no impide una vida prolongada, y su causa se desconoce.
Síndrome urémico-hemolítico. Síndrome de insuficiencia renal aguda del lactante asociada a hemólisis alteraciones de la coagulación y hemorragias. Es una afección grave de la primera infancia y se caracteriza por la presencia de hemólisis intravascular, trombocitopenia, hemorragias de la piel y las mucosas y manifestaciones neurológicas, con fenómenos de coagulación intravascular e insuficiencia renal aguda. La causa no ha sido establecida con precisión, pero se lo atribuye a una reacción atípica del organismo en respuesta a distintas infecciones por virus.
Síndrome del vaciamiento rápido. Síndrome de los gastrectomizados. Aparición de episodios posprandiales de nauseas, traspiración, palpitaciones, debilidad acentuada, sensación de calor en oleadas y a veces despeños diarreicos, en individuos sometidos a una gastrectomía parcial con gastroyeyunostomía. Se lo atribuye a un vaciamiento excesivamente rápido del contenido gástrico en el yeyuno.
Síndrome vagal del infarto del miocardio. Síndrome de los infartos de la cara inferior del corazón. El cuadro incluye trastornos, digestivos funcionales (hipo, eructos, náuseas o vómitos) fenómenos vasomotores (palidez, sudor, hipotensión arterial, lipotimia) y alteraciones del ritmo cardíaco (bradicardia sinusal o nodal, bloqueo, sinoauricular, disociación auriculoventricular con interferencias); en pacientes con infarto de miocardio de la cara inferior. Se lo observa con relativa frecuencia y responde rápidamente a la administración de atropina.
Síndrome de van Buchem. Síndrome de la hiperostosis cortical generalizada. Afección hereditaria caracterizada por osteoesclerosis del cráneo, maxilar inferior, clavículas, costillas y diáfisis de los huesos largos, con aumento de la fosfatasa alcalina sérica. Debido a la compresión progresiva por el hueso suelen producirse atrofia óptica, parálisis facial y sordera de percepción. Se trasmite como rasgo autosómico, recesivo.
Síndrome de la vena cava inferior. Síndrome de la obstrucción de la vena cava inferior. Cuadro progresivo de edema de las extremidades inferiores y del escroto, dilatación de las venas superficiales de las piernas y de la porción inferior del abdomen, oliguria y a veces ascitis. Se lo observa en cuadros graves, como la peritonitis generalizada y las trombosis ascendentes de las venas de los miembros inferiores y suele ser una complicación terminal.
Síndrome de la vena cava superior. Síndrome de la obstrucción del flujo de la vena cava superior. Se caracteriza por edema de la cara, el cuello, la porción superior del tórax y los miembros superiores (edema en esclavina), ingurgitación fija de las venas del cuello, cianosis predominantemente facial y hepatomegalia dolorosa. Obedece a compresión extrínseca de las paredes del vaso habitualmente de causa tumoral.
Síndrome de Verner-Morrison. ver Síndrome WDHA.
Síndrome de Vernet. Síndrome neurológico de la base del cráneo. Se caracteriza por parálisis unilateral progresiva y simultánea, aunque de grado variable, de los nervios craneales IX, X y XI, y obedece a la compresión ejercida por neurinomas, focos de sarcoidosis o tumores infiltrantes de la base del cráneo.
Síndrome del vértice del peñasco. ver Síndrome de Gradenigo.
Síndrome del vértigo cervical. Síndrome neurológico por insuficiencia de la circulación vertebral. Episodios transitorios, bruscos, de mareo, vértigo, nistagmo, ataxia y diplopía, o combinaciones de algunos de estos síntomas, que suelen ser desencadenados por movimientos forzados, o exageradamente amplios, de la cabeza y el cuello. Es causado por compresión o espasmo de la arteria vertebral
Síndrome de Villaret. Síndrome de la lesión de los nervios craneanos IX, X, XI y XII, y del simpático cervical, por compresión tumoral. Es unilateral y se caracteriza por: a) pérdida del gusto en el tercio posterior de la lengua (lesión del IX nervio craneano ); b) parálisis del constrictor superior de la faringe, con dificultad para deglutir sólidos (X nervio); c) parálisis del paladar blando y de las fauces, con anestesia asociada (X nervio); d) parálisis de la cuerda vocal (X nervio); e) parálisis del trapecio y el esternocleidomastoideo (XI nervio); f) parálisis de la mitad de la lengua (XII nervio), y g) síndrome de Horner (parálisis del simpático cervical). Lo producen los tumores de la parótida y del cuerpo carotídeo, y las adenopatías tumorales o tuberculosas.
Síndrome de virilización suprarrenal en el adulto. Variedad del síndrome adrenogenital. Sus manifestaciones, en la mujer, son las propias de la producción excesiva de andrógenos: hirsutismo, acné, seborrea, calvicie temporal, aumento de la masa y la fuerza muscular, amenorrea con atrofia uterina e hipertrofia del clítoris, masculinización de la voz, etc. En el varón adulto, obviamente, el cuadro es más difícil de detectar. La causa es a menudo tumoral (adenomas o carcinomas suprarrenales) y existe un aumento notable en la excreción urinaria de 17-cetosteroides.
Síndrome de Vogt. Síndrome de las lesiones del núcleo caudado y el putamen. Se caracteriza por la presencia de movimientos atetósicos, oscilación rítmica de los miembros, y llanto y risa espasmódicos y explosivos. No hay parálisis ni fenómenos sensitivos.
Síndrome de Vogt-Koyanagi. Síndrome caracterizado por alteraciones cutáneas y de los órganos de los sentidos. Es una asociación de vitiligo y áreas de alopecia, con trastornos oculares y sordera, a veces acompañada de laberintitis.
Síndrome de Volkmann. Síndrome de necrosis y retracción muscular por isquemia regional aguda. Es un cuadro isquémico, por interrupción del flujo arterial relacionado directamente con la compresión ejercida por focos de fracturas, o por los yesos confeccionados para inmovilizarlos, que habitualmente afecta la mano y la superficie flexora del antebrazo y se manifiesta por dolor intenso, tumefacción, cianosis, parestesias e imposibilidad de mover los dedos, y de no mediar una rápida resolución, por necrosis muscular y retracción cicatrizal en flexión.
Síndrome de Waardenburg. Síndrome hereditario de individuos con sordera y anomalías predominantemente faciales. Se observa albinismo de las pestañas y un mechón de cabello blanco en la región frontal, unión de las cejas en la línea media, heterocromía del iris, nariz achatada y sordera congénita. Se trasmite como un rasgo autosómico dominante.
Síndrome de Wallenberg. Síndrome neurológico del tronco cerebral con trastornos sensitivos y cerebelosos y compromiso de los nervios craneales V, IX, X y XI. Se caracteriza, del lado de la lesión, por anestesia corneal y parálisis de los músculos masticatorios (lesión del V nervio); dificultad para la deglución y pérdida del gusto en el tercio posterior de la lengua (IX nervio); parálisis del velo del paladar y de la cuerda vocal (X nervio); parálisis del trapecio y el esternocleidomastoideo (Xl nervio); síndrome de Horner (lesión de la vía descendente pupilodilatadora), y ataxia cerebelosa (vías espinocerebelosa y olivocerebelosa); y del lado opuesto por pérdida de la sensibilidad térmica y al dolor (haz espinotalámico lateral). El cuadro es producido por la oclusión de la arteria cerebelosa posteroinferior.
Síndrome de Waterhouse-Friderichsen. Síndrome de la forma fulminante de la meningitis cerebroespinal. Es un cuadro de comienzo súbito y rápida evolución, caracterizado por palidez, disnea, cianosis, fiebre elevada con escalofríos, petequias y equimosis en la piel y las mucosas, colapso y coma. Se lo observa en pacientes pediátricos y es consecuencia de una sepsis gravísima habitualmente por meningococos o neumococos.
Síndrome WDHA. Síndrome de los tumores pancreáticos productores de diarrea. El nombre deriva de las siglas; inglesas de sus manifestaciones características: diarrea acuosa (watery diarrhea), hipopotasemia (hypokaliemia) y aclorhídrica (aclorhydria). La diarrea suele ser profusa, suficientemente intensa como para llevar a veces al shock, o para producir una hipopotasemia alarmante; se comprueba una disminución de la secreción basal de ácido gástrico y a menudo hay hipercalcemia e hiperglucemia. El cuadro es producido por tumores no insulínicos de los islotes de Langerhams y sus manifestaciones obedecen, aparentemente, a la hipersecreción de polipéptidos intestinales vasoactivos.
Síndrome de Weber. Síndrome neurológico del tronco cerebral con hemiplejía alterna y compromiso del III nervio craneano. El cuadro es el de una hemiplejía del lado opuesto al de la lesión, con ptosis palpebral, estrabismo y pérdida de los reflejos a la luz y la acomodación del lado de la lesión. Es propio de las lesiones, casi siempre vasculares, que afectan el haz piramidal a nivel del pie del pedúnculo cerebral y de las fibras radiculares del III nervio craneano.
Síndrome de Weber-Christian. Síndrome de la paniculitis nodular no supurativa. Se caracteriza por la formación de nódulos dolorosos recidivantes en el tejido graso subcutáneo, con fiebre y alteración del estado general; los nódulos evolucionan dejando una depresión central. El pronóstico es benigno y la causa desconocida.
Síndrome de Wegener. Síndrome de la granulomatosis respiratoria necrotizante. Se caracteriza por una vasculitis necrotizante e infección granulomatosa de las vías respiratorias y los riñones, y a menudo de otros órganos. Existe cefalea, fiebre, artralgias, sinusitis, otitis, tos, hemoptisis, etc. Predomina en la edad media de la vida y se la considera relacionada con fenómenos de hipersensibilidad.
Síndrome de Weill-Marchesani. Síndrome congénito de distrofia mesodérmica. Se caracteriza por: a) baja estatura con braquicefalia y braquidactilia; b) tórax amplio con masas; musculares desarrolladas que dan la impresión de una gran fortaleza corporal; c) alteraciones oculares, con ectopia del cristalino y a menudo glaucoma y miopía; d) marcada rigidez articular, y ocasionalmente e) estenosis pulmonar congénita. Se trasmite como un rasgo autosómico recesivo.
Síndrome de Werdnig-Hoffmann. Síndrome de la artrofia muscular espinal familiar. Se caracteriza por la instalación temprana (primera infancia) de hipotonía y atrofia muscular, seguida de parálisis fláccida completa y finalmente la muerte. Obedece a una degeneración de las células del asta anterior de la médula y se trasmite como rasgo autosómico recesivo.
Síndrome de Wermer. Tipo 1 del síndrome de neoplasias endocrinas múltiples. Se define por la presencia de tumores o hiperplasia de las paratiroides, la tiroides, las glándulas suprarrenales, los islotes de Langerhans y la hipófisis, y por un cuadro clínico que depende de cuáles sean los sistemas; hiperfuncionantes en el momento en que el individuo consulta. Pueden observarse combinaciones de los siguientes cuadros: a) hipoglucemia; b) hipercalcemia con nefrocalcinosis; c) úlceras pépticas múltiples (síndrome de Zollinger-Ellison); d) lipomas múltiples de la piel; e) trastornos producidos por expansión de un tumor hipofisario o por disfunción de esta glándula (cefalea, pérdida gradual de la visión, amenorrea secundaria, etc.), y con menos frecuencia, un síndrome de Cushing, hepatomegalia y otras manifestaciones.
Síndrome de Werner. Síndrome hereditario con alteraciones cutáneas y tendencia al desarrollo de procesos malignos. Se caracteriza por envejecimiento prematuro, cataratas, alteraciones del tipo de las de la esclerodermia, ulceraciones en las piernas y aterosclerosis, con una tendencia aumentada al desarrollo de sarcomas y meningiomas.
Síndrome de Wernicke. Síndrome de la presbiofrenia. Se caracteriza por debilidad mental con ausencia de fijación y desorientación, angustias, alucinaciones, ideas delirantes y otros trastornos. Es una psicopatía propia de la vejez.
Síndrome de Weyers-Thier. Síndrome de la displasia oculovertebral. Se caracteriza por anomalías oculares múltiples (microftalmía, anoftalmía, etc.), asimetría facial hipoplasia del frontal y displasia orbitaria, y malformaciones del raquis y de la inserción de las costillas. Obedece a un trastorno temprano en el desarrollo de embrión y afecta solamente a los varones.
Síndrome de Wilfred Harris. Síndrome de irritaci6n del IX nervio craneal. Accesos intensos de dolor paroxístico localizado en la garganta y a veces irradiado al oído. Se repite con una periodicidad variable en ocasiones sin motivo aparente y otras; desencadenado por la deglución. La etiología es desconocida, aunque desde el punto de vista clínico el cuadro es en todo equiparable al de la neuralgia del trigémino.
Síndrome de Wilson-Mikity. Síndrome clínico y radiológico de la inmadurez pulmonar. Es la asociación de un cuadro de cianosis persistente en el recién nacido, con manifestaciones radiológicas características: focos múltiples de enfisema localizado, de 1 a 10 mm de diámetro rodeados de una banda de condensación. Otros rasgos son su presentación en prematuros, de muy bajo peso al nacer, la ausencia de signos de infección, su curso lento, la elevada mortalidad, y la posibilidad de una recuperación completa en los que sobreviven.
Síndrome de Wolff-Parkinson-White. Síndrome electrocardiográfico y clínico de excitaci6n auriculoventricular anómala. Se define por la presencia, en el electrocardiograma de una onda P normal, un PR corto (menor de 0,11 seg) y un QRS ensanchado con emplastamiento de su porción inicial (onda delta) anomalía que se asocia a una tendencia franca al desarrollo de episodios de taquicardia paroxística auricular o de fibrilación auricular. Se lo observa en sujetos jóvenes sin otros signos de cardiopatía, y obedece a la existencia de vías de conducción accesorias paralelas al haz de His.
Síndrome de Woltman-Moersch. Síndrome tetaniforme de causa no infecciosa. Se lo observa en la edad adulta, sin distinción de sexos, y se caracteriza por: a) aparición de contracciones espasmódicas intermitentes, de distribución irregular, que predominan en los músculos del tronco y las extremidades; b) estacionamiento durante períodos prolongados en estas condiciones, o progresión lenta con aumento de los grupos musculares afectados y mayor duración de las contracciones, y c) en este último caso, evolución hacia un estado de virtual rigidez generalizada. La causa es desconocida, aunque se atribuye a una deficiencia adquirida de los mediadores fisiológicos que inhiben la actividad de las motoneuronas.
Síndrome de Wunderlich. Síndrome del hematoma perirrenal espontáneo. Se manifiesta bruscamente por intensa lumbalgia unilateral, signos de anemia aguda y aparición rápida de un tumor lumbar palpable, además hay náuseas y vómitos, meteorismo por íleo reflejo, contractura lumbar y oliguria. Puede aparecer en el curso de afecciones renales (pielonefritis, tumores, etc.), hemáticas (hemofilia, leucemia) o vasculares (periarteritis, trombosis de la cava, etc.).
Síndrome de Zollinger-Ellison. Síndrome de los tumores pancreáticos ulcerógenos. Se manifiesta por una tríada característica: úlceras pépticas intratables, a veces fulminantes, hiperacidez gástrica extrema, y diarrea acuosa o esteatorrea. Es causado por la presencia de tumores no beta de los islotes de Langerhans, únicos o múltiples, benignos o malignos, y en la actualidad se lo considera dentro del tipo I de las neoplasias endocrinas múltiples.