
 Español
Español  Português
Português  English
English Dr. Julio Libman, Astrid Libman, Gaspar López Cescato e Ignacio Cadabal Muñoz
Se define la acromegalia como el cuadro clínico resultante de la exposición de los tejidos de un individuo adulto a niveles patológicamente elevados, de somatotrofina u hormona de crecimiento (GH). Cuando la hipersecreción se produce antes de la osificación de los cartílagos diafisoepifisarios de los huesos largos, lo cual permite un mayor crecimiento longitudinal, se produce el gigantismo, de observación muy poco frecuente.
La prevalencia descripta es de 39 a 69 casos por millón de habitantes, con una incidencia, de 3 a 4 nuevos casos por millón por año. No obstante, reportes europeos recientes revelan una prevalencia mas alta a la esperada, 130 casos por millón de habitantes y 86 casos por millón, donde además la polución ambiental podría estar siendo un factor predisponente
Fisiopatología
La hipersecreción de GH es producida por adenomas hipofisarios compuestos por células somatotropas.
La GH es un polipéptido de 191 aminoácidos segregado por el lóbulo anterior de la hipófisis, que estimula el crecimiento y la mitosis celular, dando por resultado un mayor tamaño y mayor número de células; desde el punto de vista clínico, esto se traduce -cuando la hipersecreción patológica ocurre después de la pubertad y de la osificación de los cartílagos diafisoepifisarios- en un aumento de la aposición ósea perióstica, con ensanchamiento y deformidad ósea, visceromegalia y aumento de los tejidos blandos.
En condiciones normales la producción de GH es afectada por varios factores fisiológicos, que ejercen su acción a través de dos hormonas hipotalámicas de naturaleza polipeptídica que llegan a los somatotropos a través del sistema portal hipotálamo-hipofisario: una estimulante, la hormona hipotalámica liberadora de GH, y otra inhibidora, la somatostatina. Las células peptidérgicas productoras de ambas se encuentran a su vez bajo el control de centros superiores por medio de diversos neurotrasmisores (norepinefrina, dopamina, serotonina). La L-dopa, previa transformación en dopamina, y los precursores de la serotonina, estimulan la secreción de GH. El bloqueo alfa-adrenérgico potencia y el beta-adrenérgico disminuye el efecto de diferentes estímulos de la secreción de GH. Estos son numerosos y variados. La hipoglucemia, el descenso de los ácidos grasos libres y el aumento de los aminoácidos circulantes estimulan la liberación de GH, mientras que la hiperglucemia y el aumento de los ácidos grasos libres la inhiben. El trauma quirúrgico y otras situaciones de estrés, así como las primeras fases del sueño que coinciden con el período de REM, se asocian con aumentos de la GH.
La GH es una hormona anabólica que tiende a favorecer la síntesis de proteínas a partir de los aminoácidos, de DNA y RNA. Tiene acción hiperglucemiante por aumento de la resistencia periférica a la insulina, lo cual determina una hiperinsulinemia que, si no es suficiente para compensar la acción de la GH, es causa de la aparición de intolerancia a los hidratos de carbono o de diabetes franca en un número apreciable de pacientes con acromegalia. Posee igualmente acción lipolítica, produciendo la hidrólisis de los triglicéridos con aumento de los ácidos grasos libres. En relación con sus propiedades promotoras del crecimiento, la GH influye en el metabolismo de diversos electrólitos, disminuyendo la eliminación urinaria de Na, K, Cl y P. El agua se retiene proporcionalmente al Na.
La GH ejerce muchas de sus acciones, excepto la hiperglucemiante y la lipolítica, estimulando la síntesis hepática y en otros tejidos de un grupo de péptidos conocidos como somatomedinas o factores de crecimiento insulinosímiles.
Si bien la acromegalia se asocia a la presencia de adenomas productores de GH, existe evidencia en estos pacientes de una alteración en el control hipotalámico sobre la secreción de GH. Se observa la liberación anómala de GH en respuesta a la administración de TRH, la falta del pico nocturno asociado al sueño, una respuesta paradójica (inhibición) a la L-dopa y la falta de supresión en respuesta a la administración de glucosa, con la aparición eventual de un incremento en vez de la habitual disminución de los niveles de GH.
Se han descrito algunos casos de acromegalia debidos a tumores hipotalámicos productores de GHRH, así como a la producción ectópica de ésta por parte de tumores del páncreas endocrino y carcinoides de pulmón; la GHRH causa en estas circunstancias una hiperplasia de los somatotropos hipofisarios.
Síntomas y signos
Existen dos tipos de manifestaciones clínicas: las determinadas por la mayor producción de GH y las alteraciones variables de las otras hormonas anterohipofisarias, por una parte, y las dependientes de la presencia del adenoma propiamente dicho, por la otra.
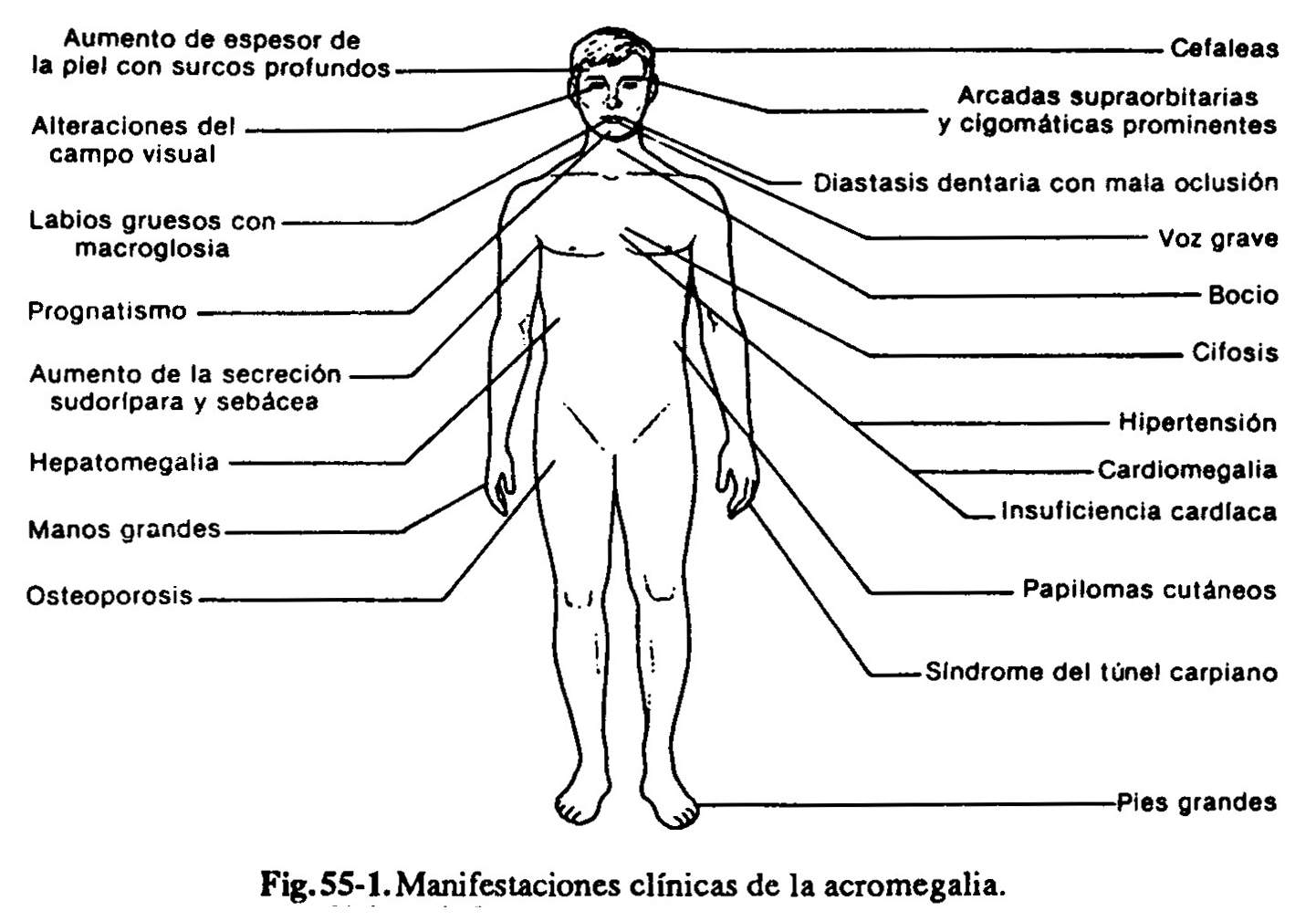
Manifestaciones endocrinas (fig. 60-1). La interpretación de los signos y síntomas producidos por la secreción exagerada de GH requiere un alto índice de sospecha, dado que suele ser confuso el límite entre las características físicas heredadas o familiares con rasgos acentuados, y los cambios similares inducidos por la acromegalia. Los pacientes refieren habitualmente una historia de cefalea, agrandamiento de las partes acrales, manos y pies, que ocurre luego del cese del crecimiento longitudinal, que se traducen por la imposibilidad de usar el mismo anillo, y la necesidad de aumentar el número de guantes y zapatos. La excesiva aposición de tejidos blandos contribuye a producir manos suculentas, con dedos en salchichón, y a aumentar el espesor del talón. Los cambios faciales suelen ser prominentes e incluyen aumento de la mandíbula (prognatismo) y del espacio entre los dientes por separación de los alvéolos de implantación (diástasis dentaria), aumento de tamaño de los huesos malares y de la nariz, prominencia de la frente (con engrosamiento de la piel, que produce pliegues transversales profundos) y agrandamiento de los labios y la lengua. Dado que el cuadro puede desarrollarse en forma insidiosa y pasar desapercibido en el curso de los años, el médico no debe aceptar de inmediato las afirmaciones del paciente y familiares de “haber tenido siempre manos y pies grandes” o de atribuir los cambios a la edad o la obesidad. La observación de fotografías antiguas obtenidas en el curso de los años puede poner de manifiesto los cambios faciales y de las manos. La voz es grave y cavernosa, por alteración de las cuerdas vocales y de las cavidades de resonancia. Los pacientes manifiestan frecuentemente excesiva transpiración, índice de hipermetabolismo, y secreción sebácea aumentada, con visualización de los orificios de desembocadura de las glándulas cutáneas en forma de puntos negros, lo cual, unido al aumento del espesor de la piel, le da un aspecto de “piel de naranja”. La existencia de excesiva transpiración y secreción sebácea es un indicio clínico de actividad de la enfermedad, entendiéndose como tal la continua producción exagerada de GH. Pueden desarrollarse pequeños fibromas cutáneos (fibroma molluscum).
Casi todos los órganos aumentan de tamaño (visceromegalia) bajo el estímulo crónico con GH. Habitualmente hay algún aumento del diámetro torácico anteroposterior, que refleja un aumento del volumen pulmonar (neumomegalia). Uno de los órganos más afectados en este sentido es el corazón. Una proporción grande de estos pacientes fallece por arritmias e insuficiencia cardiaca, a la que contribuye la propia GH a través de una cardiomiopatía específica, la hipertensión concomitante, muy frecuente y de patogenia poco clara, y la intolerancia a los hidratos de carbono o diabetes franca que se desarrolla como consecuencia de la acción diabetógena de la GH. La misma hiperinsulinemia que se desarrolla en respuesta a la resistencia periférica aumentada a la insulina contribuiría al desarrollo de aterogénesis coronaria. La cuarta parte de los pacientes presenta bocio.
Pueden existir alteraciones articulares producidas por aposición ósea exagerada y/o espesamiento cartilaginoso y sinovial, con manifestaciones de osteoartritis, dolor y limitación de movimientos. Los cambios artrósicos de la columna pueden originar diversos síndromes de compresión radicular. Pueden afectarse los nervios periféricos, por desarrollo, por ejemplo, de un síndrome del túnel carpiano, con compresión del nervio mediano, y debilidad y alteraciones sensoriales en las manos. En un comienzo es dable observar a veces un desarrollo muscular prominente, seguido de miopatía.
La aparición de astenia, fatiga, disminución de la libido e impotencia son consecuencia combinada del exceso de GH, y la falta de LH y FSH; hay miopatía, neuropatía y grados variables de hipopituitarismo por lesión del tejido hipofisario extratumoral. La oligohipomenorrea o la amenorrea son debidas a una insuficiencia ovárica secundaria al déficit de gonadotrofinas. La galactorrea obedece a la hipersecreción concomitante de prolactina por parte de células acidófilas diferentes de las productoras de GH.
Ocasionalmente, la acromegalia puede presentarse como parte del síndrome de neoplasias endocrinas múltiples tipo I, asociada a tumores de paratiroides y del páncreas endocrino.
Manifestaciones del adenoma hipofisario. La mayoría de los pacientes tienen un adenoma demostrable en el momento de la detección clínica de la enfermedad, que se manifiesta por cefaleas y/ o alteraciones visuales.
Metodología de estudio
Comprende dos partes: la evaluación endocrinológica, destinada a revelar la existencia de la hipersecreción anormal de GH, y el estudio neurorradiológico, que demuestra la existencia y el grado de extensión del adenoma hipofisario.
Evaluación endocrinológica.
Somatotrofina basal. Considerando que la GH, al igual que otras hormonas hipofisarias, se segrega en forma episódica, es conveniente obtener tres o cuatro muestras de plasma para su determinación, con intervalos de 30 a 60 minutos en el curso de un día, o una muestra diaria durante varios días consecutivos, para tener una apreciación de su concentración plasmática media. Normalmente está por debajo de 10 ng/ml; en un paciente con acromegalia puede oscilar entre esta cifra y 500 ng/ml, aunque en general se mantiene por debajo de 100 ng.
Prueba oral de tolerancia glúcida (PTOG). En los individuos normales existe una relación inversa entre glucemia y niveles de GH. Así, en el curso de una prueba oral de tolerancia glúcida, administrando 75 gramos de glucosa disueltos en 350 ml de agua, y obteniendo muestras de sangre antes y a los 60 y 120 minutos de la ingesta, se observa que las concentraciones plasmáticas de la hormona descienden a niveles muy bajos, en ocasiones no detectables.
En pacientes con niveles de IGF-I elevados se recomienda realizar la PTOG con la determinación de la hormona de crecimiento en tiempo basal y cada 30 minutos durante dos horas. La falta de supresión, en alguno de los tiempos de la prueba, a menos de 1 ng/ml o menos de 0.4 ng/ml, según el ensayo utilizado, confirma el diagnóstico . En pacientes con características clínicas de acromegalia con niveles de GH normales o dudosos, también se debe realizar la PTOG.
Cuando el estudio se lleva a cabo en pacientes acromegálicos, aproximadamente un 60% no muestra variación alguna, un 25% muestra una disminución parcial y un 15% presenta un aumento paradójico. Respuestas cualitativamente anormales pueden ocurrir en pacientes urémicos, con insuficiencia hepática, depresión, anorexia nerviosa y cáncer metastático diseminado.
Prueba de TRH. Aproximadamente el 70% de los pacientes acromegálicos presentan un aumento de la GH en respuesta a la administración de TRH, que no se observa en individuos normales.
Prueba de L-dopa. A diferencia del incremento que se produce en los normales, los pacientes acromegálicos muestran una disminución de la GH en respuesta a la L-dopa u otros agonistas dopaminérgicos.
Determinacion de IGF-I. En ausencia de enfermedades graves intercurrentes o de un cuadro de desnutrición severa los niveles séricos de IGF-I están elevados prácticamente siempre en los pacientes con acromegalia. Igualmente están aumentadas las concentraciones de la proteína ligadora de IGF (IGFBP-3).
Además, se sugiere el cálculo de la puntuación de desvío estándar o puntuación Z para expresar el número de desviaciones estándar o puntuaciones Z en que la concentración de IGF-I se sitúa por encima o por debajo de la media o la mediana de la población de referencia.
Otros estudios. El estudio radiológico de manos y pies no es necesario para el diagnóstico, pero ofrece información útil sobre las alteraciones óseas y el aumento de espesor de los tejidos blandos.
Evaluación neurorradiológica.
Radiografía simple de cráneo. De frente y de perfil, y focalizada en la silla turca, muestra en la mayoría de los pacientes una silla turca aumentada de tamaño en el momento del diagnóstico. La combinación de silla turca patológica y un seno frontal agrandado sugiere el diagnóstico.
Campimetría. Detecta las alteraciones del campo visual, que expresan la compresión de la vía óptica por el adenoma.
Tomografia axial computada y resonancia nuclear magnética. Permiten determinar la existencia de adenomas mayores de 3 mm y el grado de extensión supraselar, en el seno cavernoso o en el interior del seno esfenoidal. En la acromegalia, la conducta terapéutica se basa en la información de la RM. La tomografía computada (TC) debe permanecer en un segundo plano y ser utilizada de manera excepcional para complementar la RM en caso de malformaciones óseas, variantes anatómicas y calcificaciones.
Según el tamaño, los adenomas pituitarios se pueden clasificar en microadenomas (menores de 10 mm) y macroadenomas (mayores de 10 mm). Las características de ambos en la RM se describen a continuación:
MICROADENOMAS (secuencias de imágenes en T1 y T2)
- T1: son generalmente lesiones intraselares redondas u ovaladas, en la mayoría de los casos con señal hipointensa (en relación con la glándula normal). En aproximadamente 25% de los casos, la señal es similar a la de la glándula normal.
- T2: pueden ser hipointensos, isointensos o hiperintensos en relación a la glándula normal.
- T1 con gadolinio: típicamente muestra al adenoma hipointenso (sin captación), y alrededor la glándula normal hiperintensa por la captación del contraste.
- T1 con gadolinio (secuencias tardías): las imágenes a los 30-40 minutos luego de la inyección de contraste muestran en general la captación tardía del adenoma.
MACROADENOMAS (secuencias de imágenes en T1 y T2)
- T1: presentan una intensidad mayor que los microadenomas.
- T2: se presentan en general como lesiones heterogéneas; con algunas áreas de hiperintensidad, las cuales pueden reflejar porciones quísticas o necróticas del adenoma. En lesiones con gran extensión supraselar, esta secuencia es buena para observar el quiasma óptico, ya que el mismo es claramente hipointenso.
- T1 con gadolinio: el objetivo con esta secuencia es tratar de visualizar la glándula normal. Usualmente la glándula forma una especie de “pseudo cápsula” alrededor del tumor, que capta el gadolinio fuertemente; se ubica más frecuentemente arriba o por detrás del adenoma, y en general es unilateral. El tallo hipofisario usualmente está lateralizado hacia el lado donde se encuentra la glándula.
Bibliografia:
- GUIA DE RECOMENDACIONES PARA EL DIAGNÓSTICO CLÍNICO, BIOQUÍMICO Y POR IMÁGENES DE LA ACROMEGALIA, FEDERACIÓN ARGENTINA DE SOCIEDADES DE ENDOCRINOLOGÍA (FASEN) AÑO 2017