
 Português
Português  Español
Español  English
English Juan C. Linares Casas
A doença isquêmica ou coronariana é uma doença heterogênea que abrange um amplo espectro de manifestações clínicas, desde doença assintomática - isquemia silenciosa - e angina estável, por um lado, até angina instável, infarto agudo do miocárdio, cardiomiopatia isquêmica e morte súbita, em o outro. Estima-se que 30 a 40% dos eventos coronários agudos ocorram em pessoas sem história clínica de patologia isquêmica; portanto, a prevenção de crises agudas constitui um verdadeiro desafio na medicina cardiovascular. Um conhecimento mais profundo de suas bases fisiopatológicas pode nos levar a formular estratégias de prevenção mais eficazes.
Fisiopatologia.
O substrato anatômico usual - não exclusivo - da obstrução coronariana é a redução da luz vascular por placas ateromatosas. Essas placas estão localizadas nas artérias epicárdicas e irão gerar progressivamente uma diminuição no suprimento de oxigênio abaixo de um nível crítico de demanda em uma determinada área do ventrículo esquerdo.
1.- Diminuição do fornecimento de O2: este fornecimento é controlado por fatores hidráulicos e anatômicos que regulam o fluxo coronário:
a) Os fatores hidráulicos são: pressão de perfusão coronariana, ligada por sua vez à pressão aórtica; pressão diastólica final do ventrículo esquerdo e tempo de fluxo coronário na diástole. Como se sabe, o fluxo sistólico para o miocárdio subepicárdico chega a 30% do total, mas o subendocárdio não recebe fluxo na sístole. Uma restrição ao fluxo coronário na diástole, portanto, produz uma hipoperfusão acentuada do subendocárdio.
b) Os fatores anatômicos são dados pelo leito vascular coronariano e a presença de aterosclerose obstrutiva nos grandes vasos epicárdicos. A resistência ao fluxo causada pelas estenoses ateromatosas quando ultrapassam a redução de 50% do diâmetro vascular gera um gradiente transstenótico com queda da reserva coronariana, que será maior à medida que a redução do diâmetro aumenta.
Deve-se acrescentar aqui que a rápida aceleração das lesões obstrutivas se deve à instalação de uma trombose na placa de aterosclerose, que pode levar à oclusão ou suboclusão da artéria afetada. Muitos autores chamam essa condição de aterotrombose, para enfatizar o papel transcendente dessa associação patogênica nas doenças coronárias agudas.
2.- Demanda aumentada de O2: a demanda de oxigênio é determinada pela freqüência cardíaca, duração da sístole, contratilidade miocárdica e tensão da parede ventricular esquerda.
Qualquer aumento em qualquer um desses fatores aumentará as necessidades de oxigênio, levando a um aumento no gradiente transestenótico e uma queda adicional na reserva coronariana. É o caso da estenose aórtica, hipertensão arterial e taquiarritmias.
No entanto, as síndromes coronárias agudas e crônicas são processos ativos e não se limitam a um ambiente estático de placas ateromatosas fixas. Seus agentes causadores são, na verdade, anormalidades de um ambiente molecular e biológico hostil ao endotélio vascular, ambiente que é estimulado por um sistema de sinais que alteram sua integridade, desencadeando diversas reações inflamatórias. A placa de ateroma é hoje concebida como a sede das reações bioquímicas entre o endotélio unicelular, a célula muscular lisa subjacente e os elementos do sangue que, em outro momento, circulam normalmente. Graças à compreensão dessas interações celulares e à magnitude de seus danos, podemos agora diferenciar a placa ateromatosa estável da instável ou vulnerável.
A aterosclerose é, em síntese, a consequência de uma interação entre diferentes agentes agressores (dislipidemia, tabaco, hipertensão, diabetes) e respostas defensivas muito complexas. Como resultado dessa "luta", a parede arterial sofre alterações estruturais e funcionais progressivas.
O endotélio normal e patológico.
Apesar de suas dimensões microscópicas, o endotélio vascular é um órgão endócrino de extrema importância, cuja integridade é requisito fundamental para a preservação da estrutura e função da parede vascular, bem como para a regulação dos fenômenos trombóticos. Em um homem de 70 kg, tem uma área equivalente a 6 quadras de tênis, um peso total de mais de dois quilos - mais que o fígado - e um número total de células naquela monocamada de um trilhão de unidades. Essas células recebem e emitem sinais. Eles "percebem" mudanças no fluxo, pressão, sinais inflamatórios ou níveis de hormônios ou moléculas em funcionamento e respondem sintetizando, liberando e ativando substâncias que regulam quatro funções primárias:
Tônus vascular
O crescimento e proliferação de células musculares lisas
Trombose e homeostase
Resposta inflamatória e imunológica
Como importante órgão metabólico e endócrino, atua regulando essas funções de dupla forma: secreta fatores relaxantes e constritores vasculares, trombogênicos e antitrombóticos, fibrinolíticos e antifibrinolíticos, inibidores e estimulantes do crescimento celular, etc.
Mas devemos enfatizar que, em condições normais, atua de forma inibitória, facilitando a circulação: secreta fatores vasodilatadores, inibe a contração da musculatura lisa vascular, trombose, agregação plaquetária, crescimento celular e adesão leucocitária. Mas em condições patológicas, quando o endotélio é lesado ao ser atacado por diferentes noxas, ele é “ativado” e sua função será oposta: sua superfície irá estimular a coagulação e a adesão celular, ao mesmo tempo que produz substâncias vasoconstritoras e estimulantes do crescimento celular que elas vai engrossar sua parede.
As substâncias vasorrelaxantes incluem óxido nítrico e prostaciclina, que são antiinflamatórios, inibidores do crescimento e, no caso da prostaciclina, antitrombóticos. O fator transformador de crescimento beta, a trombospondina e o sulfato de heparan também inibem o crescimento celular. Os antitrombóticos incluem uroquinase, heparina proteoglicanos, ativador do plasminogênio tecidual e trombomodulina.
Essa disfunção endotelial caracteriza o estágio inicial da aterosclerose, quando as alterações estruturais vasculares ainda não ocorreram. O equilíbrio entre os fatores endoteliais relaxantes e constritivos tem sido desequilibrado, com predomínio destes últimos: endotelina, angiotensina II, fator de crescimento derivado de plaquetas (PDGF), bem como substâncias pró-trombóticas (fator tecidual, fator ativador de plaquetas, Von Fator de Willebrand, inibidor do ativador do plasminogênio tecidual), fatores de crescimento vascular e citocinas pró-inflamatórias.
A produção deficiente ou alterada de óxido nítrico (NO) tem se mostrado amplamente responsável pela disfunção endotelial, em resposta a certos estímulos patológicos. Esses estímulos bioquímicos e biofísicos são capazes de desequilibrar a homeostase endotelial, levando ao aumento da permeabilidade das lipoproteínas aterogênicas, aumento da adesão de monócitos, estimulação na produção de citocinas e produtos finais avançados de reações biossintéticas, bem como alteração na hemodinâmica e forças biomecânicas que levam a maior dano endotelial. Tudo isso resulta em trombose, inflamação, reatividade vascular anormal, hiperplasia intimal e fibrose.
A estria gordurosa será a primeira manifestação histopatológica da doença aterosclerótica, que irá progredir até a formação da placa fibrosa, que consiste em um núcleo lipídico recoberto por uma camada fibrosa resultante da síntese de colágeno, elastina e proteoglicanos pelas células musculares. liso e macrófagos que migraram para a íntima arterial.
O conceito de Angina Pectoris
O termo "angina pectoris" ou "angor pectoris" foi introduzido por Heberden em 1768 para indicar um "distúrbio do peito" muito característico, acompanhado por "sensação de estrangulamento e ansiedade". Ao usar a palavra “angina” (estrangulamento), Heberden a descreveu como um tipo de dor no peito de uma modalidade muito especial que é acompanhada por fenômenos psíquicos, o mais notável dos quais consistia no medo da morte iminente (angor animi).
A angina pectoris é uma síndrome em si, não uma doença. Indica sempre o mesmo distúrbio fisiopatológico básico: isquemia miocárdica, qualquer que seja a causa que a gera.
Embora seja quase impossível encontrar uma definição clara de angina de peito nas publicações, pode-se estabelecer que se trata de uma síndrome clínica caracterizada por crise de dor, rigidez ou desconforto de origem isquêmica, geralmente localizada atrás do esterno, mas localizada às vezes na mandíbula, ombros, costas ou braços, agravada ou desencadeada por esforço ou emoções, e que é rapidamente aliviada quando cessa a atividade ou distúrbio causal, ou pela administração de nitroglicerina.
Alguns elementos descritos acima podem não ser típicos, mas a característica opressora da dor ou desconforto é a base para seu reconhecimento. A localização, a radiação ou a duração podem variar, podendo nem mesmo estar relacionadas aos esforços, mas a sensação opressora e constritiva de peso constitui o caráter mais preciso da angina.
Características da dor.
A localização geralmente é retroesternal, embora o local inicial às vezes seja mais amplo e inclua a maior parte da região precordial. Às vezes, não está localizado no tórax: pode afetar a mandíbula, o braço esquerdo, o epigástrio, os ombros, a região escapular e outras áreas atípicas. Nestes casos, sua aparência paroxística após o esforço, sua natureza opressiva e sua rápida cessação com repouso sugerem isso.
Embora possa estar localizada atrás do esterno, a dor tende a se irradiar para o pescoço, mandíbula e membros superiores. Em sua forma mais clássica, irradia para a escápula e parte superior do braço esquerdo. Às vezes, a distribuição do nervo ulnar ao longo do aspecto ântero-medial das mãos e dedos segue claramente.
Quanto ao seu caráter, o que foi dito é reiterado: é uma pintura classicamente constritiva ou opressora. Também foi descrito como asfixia ou queimação. Há momentos em que os pacientes não relatam dor, mas apenas aperto ou sensação de peso no peito. Pode ser leve ou intenso, mas sua qualidade é característica. Em muitos casos, a dor imita tanto a tensão atrás do esterno que o paciente tenta vomitar para obter alívio.
Geralmente força o paciente a ficar o mais imóvel possível e, se estiver marchando, deve parar. Geralmente é acompanhada, como já mencionado, por uma sensação de morte iminente, que não está relacionada à sua intensidade. À medida que as crises ocorrem com o passar do tempo, os pacientes deixam de sentir essa angústia, devido ao maior conhecimento ou experiência que adquiriram diante da doença.
Os episódios de angina geralmente começam gradualmente, atingem sua intensidade máxima logo e também desaparecem gradualmente em alguns minutos, geralmente de dois a dez. Eles são aliviados rapidamente com a cessação da atividade que os causou, repouso ou administração de nitroglicerina sublingual. Se a dor durar trinta minutos ou mais, deve-se suspeitar da instalação de um acidente coronariano agudo (angina instável ou infarto do miocárdio).
Gatilhos.
O esforço é a causa mais importante e frequente do desencadeamento da angina de peito, pois aumenta a demanda de oxigênio. O exercício físico mais comum é caminhar ou subir escadas. A gravidade da condição está relacionada ao esforço necessário para gerar a dor.
A exposição ao frio e à digestão aumentam a possibilidade de angina após o esforço, que geralmente não é seguida de dor. Da mesma forma, estados emocionais podem desencadear crises anginosas por meio de complexos mecanismos adrenérgicos.
Mas é importante observar que em muitos pacientes os episódios de dor ocorrem sem causa aparente e em repouso, em decorrência de uma queda repentina no suprimento de oxigênio ao miocárdio, em decorrência da ruptura da placa de ateroma com adição trombose (acidente coronário agudo), ou como expressão de uma obstrução coronária dinâmica (espasmo). Voltaremos a esses pontos mais tarde.
A frequência com que a angina se apresenta de forma atípica e o grande número de doenças que se apresentam com dor torácica exigem um diagnóstico diferencial cuidadoso e muitas vezes difícil. As dores pontiagudas, fugazes e localizadas no precórdio são geralmente de origem psíquica. A dor da dissecção aórtica, do pneumotórax e, em geral, dos processos que ocorrem com ruptura ou laceração dos tecidos, começa abruptamente e sua intensidade é máxima desde o início.
As circunstâncias e sintomas que acompanham a dor são muito úteis no diagnóstico diferencial. A relação com a ingestão de alimentos ou melhora com álcalis levará a um distúrbio digestivo; mudanças na intensidade com o movimento dos membros superiores ou pescoço e com a respiração ou posição corporal sugerem radiculopatia cervical ou pericardite, respectivamente. Em contraste, a dor anginosa, opressiva e angustiante costuma ser acompanhada por uma sensação de gravidade e, ocasionalmente, sudorese e palpitações. A dispneia durante a dor é rara, mas seu aparecimento indica uma doença coronariana grave e é um sinal de mau prognóstico. A presença de fatores de risco - hiperlipidemia, diabetes, hipertensão, tabagismo,
Classificação clínica da angina de peito
As circunstâncias em que a dor anginosa aparece de forma ampla indicam o mecanismo que a causa e, por sua vez, o seu conhecimento permite orientar o tratamento. Com base nesses critérios, diferentes classificações foram propostas. Consideraremos aqui: a) angina pectoris estável; b) angina de peito instável, que faz parte das síndromes coronárias agudas, ec) vasoespástica ou angina de Prinzmetal.
Angina pectoris estável.
O diagnóstico de angina pectoris estável é feito pela relação entre dor coronariana e exercício. É definida como aquela que não mudou suas características nos últimos 3 meses, com ou sem história de infarto do miocárdio. Constitui 60% dos casos diagnosticados.
Em geral, o nível de esforço necessário para causar angina, ou limiar de angina, é constante por longos períodos de tempo, de modo que o paciente geralmente sabe com antecedência quais atividades em sua vida diária irão causar isso. Quando um paciente com angina estável muda as características de sua dor - aparência mais frequente, esforços provocativos menos intensos, etc. - ele terá entrado no grupo de angina instável.
De acordo com sua gravidade e a limitação funcional que impõe ao paciente, a angina de esforço é dividida em quatro graus de acordo com a classificação da Canadian Cardiovascular Society:
Grau I : a atividade física regular não produz angina, que aparece com esforços significativos, rápidos e / ou prolongados.
Grau II : limitação leve de atividade física; a dor surge ao caminhar em ritmo normal por dois ou três quarteirões ou subir mais de um andar rapidamente, caminhar em encostas, no período pós-prandial, ou em climas frios ou contra o vento; em estados emocionais ou ao iniciar atividades matinais.
Grau III : limitação significativa da atividade usual: a dor ocorre ao subir no chão ou andar em um bloco com uma marcha normal.
Grau IV : incapacidade de realizar qualquer atividade física sem o aparecimento de angina; pode ocorrer angina em repouso.
Quase 80% dos pacientes com angina estável pertencem às classes I e II. Pacientes de grau III-IV mostraram ter mortalidade mais alta do que aqueles de graus I e II.
O exame físico costuma ser normal, especialmente após o término da convulsão. Durante o acesso anginoso, podem aparecer palidez, diaforese, taquicardia e ausculta de um quarto ruído. A presença de hipotensão e insuficiência cardíaca durante os ataques são sinais de gravidade.
O eletrocardiograma é normal em 50% dos casos, na ausência de dor. O restante pode mostrar sinais de um infarto antigo, hipertrofia ventricular esquerda, depressão do segmento ST ou alterações isquêmicas na onda T.
O teste ergométrico constitui um teste diagnóstico muito importante nesses pacientes. É considerado positivo se durante o mesmo ou imediatamente após o aparecimento de angina ou se o segmento ST desce pelo menos 1 mm adotando uma forma horizontal. Se a dor aparecer precocemente (menos de 6 minutos) ou se a depressão do segmento ST exceder 2 mm ou a pressão arterial cair durante o esforço, estaremos lidando com um paciente de alto risco que deve ser encaminhado para estudo angiográfico coronariano com vistas a revascularização.
Os estudos de imagem radioisotópica e a ecocardiografia sob estresse podem identificar a extensão, gravidade e localização da isquemia. A angiografia coronária e a tomografia multislice nos informarão sobre o estado da função ventricular e a extensão das lesões coronárias. É importante destacar que a sobrevida está relacionada ao grau da aterosclerose coronariana: quanto maior o número de artérias coronárias obstruídas e quanto maior a deterioração contrátil do ventrículo esquerdo, pior o prognóstico. Nesse sentido, os achados da cineangiocoronariografia são os melhores fatores preditivos.
Em relação à morfologia coronariana na angina estável, deve-se ressaltar que as imagens das placas de ateroma são as de um ateroma não complicado, ou seja, com superfície lisa, branco-amarelada, sem ulcerações, hemorragias intimais ou trombos aderentes. Nessa indenização da cobertura fibrosa da placa está a diferença com as lesões do tipo instável.
Angina pectoris instável.
Alguns tipos de angina de peito são considerados formas instáveis de doença coronariana, e a abordagem terapêutica difere significativamente da angina estável, pois sua presença indica uma situação de evolução imprevisível. Sob o termo angina instável podemos incluir os seguintes tipos: a) angina de início recente (duração dos sintomas menos de um mês); b) angina progressiva: é aquela em que as crises dolorosas se tornam mais frequentes ou de maior duração, são rebeldes à nitroglicerina ou surgem com esforços cada vez menos intensos; c) o terceiro subgrupo é constituído pela angina em repouso.
A angina instável faz parte do chamado grupo das síndromes coronarianas agudas, para as quais será analisada posteriormente.
Angina de Prinzmetal.
A angina de Prinzmetal ou angina variante é uma manifestação clínica particular de doença isquêmica do coração caracterizada por ataques dolorosos em repouso e elevação do segmento ST durante eles (às vezes na forma de uma onda monofásica).
Essa condição responde a um aumento do tônus vasomotor coronário ou a um espasmo no vaso, que pode se instalar em uma artéria coronária saudável ou ateromatosa e que causará isquemia por meio de uma diminuição no suprimento de oxigênio.
O eletrocardiograma geralmente é normal fora das crises de angina, especialmente na ausência de aterosclerose coronariana.
Pacientes com espasmo coronariano apresentam resposta muito acentuada à administração de ergonovina, que reproduz o quadro clínico e eletrocardiográfico da angina variante. É, no entanto, um teste potencialmente perigoso que deve ser evitado se o estado da árvore coronária não for conhecido.
SÍNDROMES CORONÁRIOS AGUDOS
As síndromes coronárias agudas (SCA) são causa frequente de consultas na prática médica diária. A sua importância reside na elevada morbilidade e mortalidade e no elevado custo com a saúde que gera, este último ligado ao uso de medicamentos e à realização de testes paraclínicos e à realização de procedimentos diagnósticos e terapêuticos invasivos.
Definição e classificação
SCA é um termo operacional que foi desenvolvido para se referir a uma constelação de sintomas consistentes com isquemia miocárdica aguda. Isso engloba um espectro variado de entidades clínicas, desde angina instável (IA) até infarto agudo do miocárdio (IAM) em todas as suas variantes.
Os ACS são classificados de acordo com as anormalidades do segmento ST do eletrocardiograma (ECG) inicial em:
- ACS com supradesnivelamento do segmento ST; Y
- ACS sem elevação do segmento ST.
Isso se justifica pelo fato de que o primeiro grupo de pacientes será considerado infarto agudo e a estratégia de maior importância terapêutica é a reperfusão miocárdica de emergência por trombólise ou angioplastia.
No segundo grupo, apresentam-se pacientes com angina instável e IAM sem supradesnivelamento do segmento ST, diferindo entre si pela ausência ou presença, respectivamente, de marcadores biológicos e enzimáticos no sangue; da mesma forma, o manejo terapêutico inicial é o mesmo para ambas as entidades.
Fisiopatologia das síndromes coronárias agudas
Os ACS compartilham uma origem fisiopatológica comum (Fig. 1): o acidente da placa de aterosclerose, com erosão ou ruptura de seu revestimento fibroso e, como consequência de vários tipos de lesão, forças hemodinâmicas e provavelmente inflamação, expõe um substrato altamente trombogênico que Ao interagir com o sangue, gera dois eventos marcantes a) ativação e agregação plaquetária eb) geração de trombina. Ambos os eventos interagem e dão origem a um trombo de maior ou menor magnitude.
Se o trombo for oclusivo, ele irá gerar a interrupção total do fluxo coronário no vaso culpado e geralmente a consequência é infarto do miocárdio transmural ou infarto Q (com elevação do segmento ST), ao passo que se a trombose adicionada não for oclusiva, o quadro é geralmente O quadro clínico que acompanha é AIS sem supradesnivelamento de ST, ou seja, angina instável ou infarto sem Q.
A placa vulnerável ou de alto risco.
Apesar das lesões ateromatosas terem a mesma fisiopatologia, as placas são muito heterogêneas e as de "alto risco" têm características próprias. O conhecimento da doença aterosclerótica avançou da velha ideia que sustentava que a oclusão aguda de uma artéria era a conseqüência final de uma obstrução lentamente progressiva, ao conceito atual que estabelece que ela é produto da ruptura de uma placa, com posterior formação de um trombo que oclui parcial ou totalmente o lúmen arterial. Mas só recentemente foi postulado que a composição da placa é mais importante como indicador de risco de ruptura do que o grau anterior de obstrução, tornando-se o fator determinante dessa doença.
A placa aterosclerótica com tendência à ruptura, também denominada "vulnerável" ou "alto risco" possui, do ponto de vista histológico, um grande núcleo lipídico, geralmente excêntrico e com depósitos lipídicos extracelulares, alta densidade de linfócitos T e macrófagos repletos de lipídeos. , um número reduzido de células musculares lisas, e todas cobertas por uma fina camada fibrosa (fig. 1). Sua consistência é a de um creme dental à temperatura ambiente, sendo ainda menos consistente à temperatura corporal. Portanto, não é surpreendente que essas placas sejam pouco estáveis e sujeitas a ruptura, ainda mais quando comparadas a outras placas fibrosas ricas em colágeno (fig. 2).
Uma vez que a fina camada fibrosa é rompida ou erodida, seu interior lipídico fica exposto à corrente sanguínea que desencadeia a cascata de coagulação, gerando um trombo que oclui ou obstrui o lúmen, com manifestações clínicas de síndromes coronárias agudas (fig. 3).
 figura 1
figura 1
 Figura. 2: A placa estável possui uma camada fibrosa relativamente espessa que protege o núcleo lipídico do contato com o sangue.
Figura. 2: A placa estável possui uma camada fibrosa relativamente espessa que protege o núcleo lipídico do contato com o sangue.
 Figura 3: Esquematização fisiopatológica das síndromes isquêmicas agudas.
Figura 3: Esquematização fisiopatológica das síndromes isquêmicas agudas.
IAM Q: infarto do miocárdio tipo Q ou transmural; AI: angina instável;
MI não Q: infarto agudo do miocárdio tipo não Q.
A angina instável sugere que houve uma erosão ou fissura relativamente pequena de uma placa vulnerável que dá origem a episódios transitórios de oclusão trombótica com a consequente angina em repouso. Este trombo é geralmente lábil, com oclusões que não excedem 15-20 minutos. Além disso, a liberação de substâncias vasoativas pelas plaquetas causará vasoconstrição secundária, o que pode contribuir para reduzir ainda mais o fluxo coronariano.
No infarto não Q, o dano da placa é mais importante e resulta em oclusões trombóticas mais persistentes, que podem durar até uma hora. Um quarto dos pacientes com infarto não Q tem oclusão coronariana por mais de uma hora, mas o território miocárdico isquêmico geralmente é irrigado por colaterais.
No infarto do tipo Q ou transmural, ocorre uma grande fratura da placa que pode resultar na formação de um trombo fixo e persistente por mais de uma hora com a conseqüente necrose transmural do miocárdio comprometido. Alguns casos de morte súbita coronariana são provavelmente baseados em uma lesão rapidamente progressiva, onde a ruptura da placa e a trombose resultante levam a arritmias ventriculares fatais.
Clínica
Pacientes com angina instável e infarto do miocárdio freqüentemente se queixam de dor anginosa com características semelhantes às da angina estável, mas são mais graves e prolongadas. Assim como na angina de esforço clássica a dor dura entre 2 e 10 minutos, aproximadamente, na angina instável persiste entre 10 e 25 minutos, aparece com esforços menos intensos e mesmo em repouso.
No infarto do miocárdio, a dor anginosa geralmente dura mais de 30 minutos - às vezes horas - e costuma ser mais intensa. Não é aliviado pela nitroglicerina e pode ser acompanhado por arritmias ou sintomas e sinais de insuficiência cardíaca. Não se intensifica ao sentar ou respirar profundamente, um critério útil para diferenciá-lo da dor da pericardite aguda.
A maioria dos pacientes com ataque cardíaco, especialmente aqueles com elevação do segmento ST, fica angustiada e agitada e tenta, sem sucesso, aliviar a dor movendo-se na cama, modificando sua postura. É comum encontrar palidez junto com sudorese e frio nas extremidades. A combinação de dor torácica retroesternal com duração superior a 30 minutos e sudorese é um forte argumento a favor de um infarto agudo do miocárdio. Além disso, geralmente ouve-se um galope pré-sistólico (R4) ápice, devido à forte contração do átrio esquerdo devido ao aumento da pressão diastólica do ventrículo esquerdo.
A pressão arterial e a frequência cardíaca podem mostrar hipertensão e taquicardia ou hipotensão e bradicardia, dependendo se há hiperatividade simpática ou parassimpática, respectivamente, embora em infartos com elevação de ST a pressão sistólica geralmente caia em cerca de 10 ou 15 mm Hg.
Após os primeiros dias após o infarto, ocorre fricção pericárdica em 6 a 10% dos casos, que geralmente é intermitente e é ouvida mais claramente no ápice ou na borda esternal esquerda; pode persistir por vários dias e é decorrente de pericardite localizada devido à extensão transmural da necrose para o epicárdio.
A febre geralmente ocorre dentro de 24 horas após o infarto, geralmente em grau moderado e durando entre dois e quatro dias. Às vezes há vômito, às vezes por causa da medicação usada.
Em cerca de metade dos casos, um fator desencadeador é detectado antes do ataque cardíaco, em geral um exercício físico intenso e incomum, um estresse emocional ou uma doença médica ou cirúrgica. Pode ocorrer a qualquer hora do dia ou da noite, mas sua frequência atinge seu valor máximo nas primeiras horas do despertar. Esse pico circadiano se deve à combinação de aumento do tônus simpático e maior tendência à trombose - maior agregabilidade plaquetária - que ocorre entre 6h e 12h.
No caso de disfunção ventricular esquerda, choque cardiogênico e insuficiência mitral isquêmica, os sinais dessas complicações serão encontrados.
Antes de mais nada, devemos sublinhar um ponto que não devemos esquecer: cerca de 25% dos ataques cardíacos não são clinicamente reconhecidos; metade deles são assintomáticos e o diagnóstico é feito retrospectivamente por meio da análise do eletrocardiograma. No resto, a dor é atípica ou inexistente. Alguns autores fornecem porcentagens ainda mais altas de ataques cardíacos "silenciosos".
Eletrocardiograma.
O eletrocardiograma (ECG) é de suma importância e permite que a SCA seja classificada em dois grandes grupos: com e sem supradesnivelamento do segmento ST. O primeiro geralmente desenvolverá um infarto do miocárdio com onda Q (infarto transmural), enquanto o último provavelmente terá angina instável ou um infarto sem onda Q. A diferença entre as duas últimas condições será dada pela presença ou ausência de marcadores bioquímicos de necrose nos exames de sangue, como veremos mais adiante.
Enquanto uma espessura suficiente da parede ventricular esquerda é afetada, e mais especificamente nos infartos transmurais, as alterações no ECG ocorrem devido a: a) isquemia; b) lesão miocárdica mais avançada ou área denominada “lesão” ec) necrose ou próprio infarto. Essas alterações se desenvolvem de forma dinâmica e evolutiva, sucessivamente, condicionando as seguintes modificações:
Inicialmente, nos primeiros minutos de oclusão coronariana, a isquemia modifica a repolarização ventricular e produz inversão das ondas T, que aparecerão negativas, pontiagudas e profundas nas derivações que ficam de frente para a área isquêmica. Portanto, em infartos da parede anterior, os T são provavelmente negativos nas derivações precordiais; nos infartos laterais, serão em D1 e aVL; e nos infartos póstero-inferiores o T será invertido em D2, D3 e aVF.
À medida que a isquemia e o dano miocárdico consecutivo são acentuados, as chamadas "correntes de lesão" ou "lesão" aparecem nas derivações que olham para a lesão e que se traduzem no ECG por uma elevação do segmento ST, elevação que atinge sua expressão máxima no 1º ou 2º dia do evento. Essa elevação do segmento ST pode até se tornar do tipo onda monofásica (fig. 4).
A área de necrose miocárdica não gera potenciais de ação. Se o infarto cobrir toda a espessura da parede ventricular (infarto transmural), pode ser considerado uma janela aberta na parede. Portanto, o potencial negativo dentro da câmara ventricular é transmitido através do tecido necrótico para os eletrodos que ficam de frente para a área infartada. Ao invés da onda positiva normal, o início do complexo QRS ventricular será direcionado para baixo, ou seja, haverá ondas Q profundas e anormais, e apenas complexos eletrocardiográficos negativos do tipo QS. Essas ondas Q são mais largas e profundas do que as ondas Q normalmente vistas em algumas derivações. Eles aparecem várias horas após o início da imagem, sucedendo às alterações no segmento ST.Seu aparecimento tardio obriga, portanto,
A onda de lesão ou elevação positiva do segmento ST é, portanto, essencial para o diagnóstico do infarto transmural e sua extensão é proporcional ao seu tamanho ou extensão. Esse tecido lesado surge desde as primeiras horas após a oclusão e persiste, geralmente em declínio gradual, por 3 a 4 semanas, evoluindo para necrose total (ondas Q profundas) ou, pelo menos em parte, para isquemia simples, indicando neste último caso uma evolução melhor. À medida que a elevação do ST melhora, haverá um aumento da isquemia no subepicárdio (ondas T negativas).
No caso de síndromes coronarianas agudas sem supradesnivelamento do segmento ST, ou seja, angina instável e infarto sem Q.
Na angina instável, o ECG pode não apresentar alterações, mas na maioria dos casos podem ser observadas alterações transitórias: infradesnivelamento do segmento ST maior que 1 mm ou maior que 0,5 mm, inversão das ondas T ou mesmo bloqueio do segmento ST. ramo esquerdo de grau avançado e arritmias.
No infarto não Q, ocorre que o tecido necrótico não ocupa toda a espessura da parede ventricular e, portanto, a onda de ativação pode atingir a área transmural sã pelo seu trajeto usual. A “janela” elétrica de infartos Q não ocorre aqui. Podemos observar diminuição da tensão das ondas R nas derivações que olham para a zona de infarto, desvio do segmento ST, ondas T invertidas (fig. 5) e bloqueio do ramo, como na angina instável.
A diferença entre as diferentes condições coronárias agudas sem supradesnivelamento do segmento ST não deve ser encontrada no ECG, mas nos marcadores bioquímicos de necrose.
 Figura 4: ECG de um infarto agudo com elevação do segmento ST, evidente nas derivações II, III e aVF
Figura 4: ECG de um infarto agudo com elevação do segmento ST, evidente nas derivações II, III e aVF
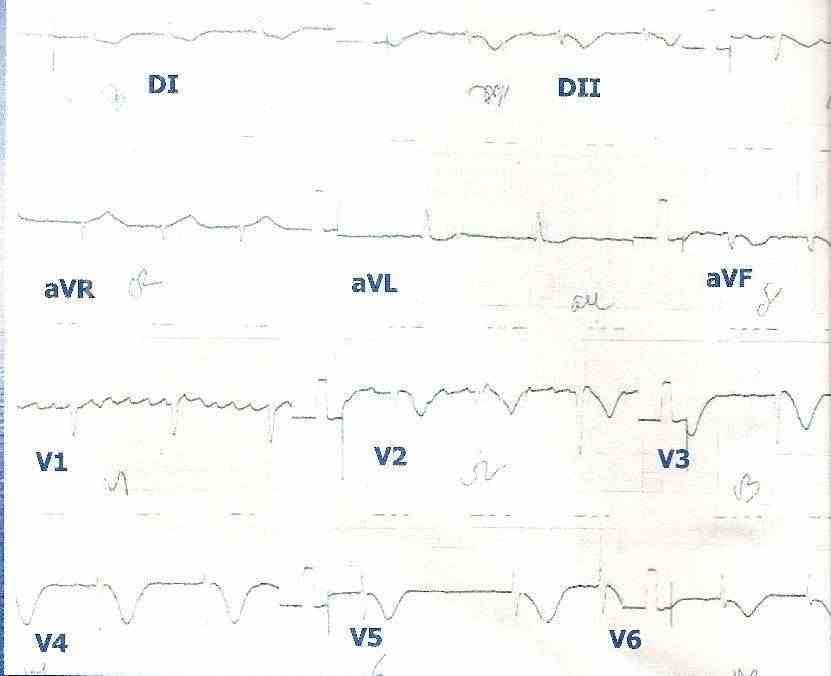 Figura 5: Síndrome coronariana aguda sem supradesnivelamento do segmento ST em um paciente com fibrilação atrial crônica. A inversão das ondas T pode ser observada nas derivações V2, V3, V4, V5 e V6.
Figura 5: Síndrome coronariana aguda sem supradesnivelamento do segmento ST em um paciente com fibrilação atrial crônica. A inversão das ondas T pode ser observada nas derivações V2, V3, V4, V5 e V6.
Marcadores bioquímicos de necrose miocárdica
O infarto do miocárdio causa várias alterações humorais, como leucocitose e aumento da velocidade de hemossedimentação. No entanto, do ponto de vista diagnóstico, apenas o aparecimento no sangue de diferentes proteínas liberadas na corrente circulatória, liberadas por miositas lesados, é importante: mioglobina, troponinas cardíacas T e I, creatina fosfoquinase (CPK), desidrogenase láctica (LDH ) e outros. . O diagnóstico de IAM é estabelecido quando os níveis hemáticos de biomarcadores específicos e sensíveis, como mioglobina, troponinas e a fração MB da CPK, estão aumentados no quadro clínico de isquemia aguda. Esses marcadores refletem o dano miocárdico, mas não indicam seu mecanismo. Portanto,
Geralmente são usados dois marcadores. O mais recomendado é usar uma combinação de um marcador de aumento rápido, como a mioglobina, e outro que leva mais tempo para subir, mas é mais específico, como as troponinas, a fim de detectar a presença de infarto em ambos os pacientes que se apresentam precocemente .
A mioglobina é detectável no sangue duas a três horas após o início. Sua concentração sobe rapidamente, atinge o nível máximo entre 6 e 12 horas após o início dos sintomas e cai para os valores normais nas 24 horas seguintes, com eliminação pelos rins.
As troponinas cardíacas são consideradas os marcadores de especificidade cardíaca mais comumente usados no diagnóstico de lesão miocárdica, particularmente a troponina I e a troponina T. Essas proteínas estão associadas a sequências de aminoácidos específicas codificadas por genes diferentes daqueles que codificam isoformas do músculo esquelético. Assim, a TnI tem especificidade completa para o miocárdio, enquanto a TnT ocorre em pequenas quantidades no músculo esquelético durante o desenvolvimento fetal humano e é reexpressa em patologias associadas à regeneração muscular (por exemplo, distrofia muscular de Duchenne). As troponinas aparecem no soro relativamente cedo após o início do infarto (4 a 10 horas), atingem seu pico em 48 horas e permanecem anormais por 4 a 10 dias.
Se a determinação da troponina não estiver disponível, a melhor alternativa é CPK-MB: é menos específica do que a troponina cardíaca, mas sua especificidade clínica para lesão irreversível é mais robusta. Tal como acontece com as troponinas, um aumento de CPK-MB (por exemplo, para o diagnóstico de IAM) é aquele que excede 99% dos valores de um grupo de controle.
As medidas de CPK total geralmente não são recomendadas para o diagnóstico de rotina de infarto, devido à ampla distribuição dessa enzima nos tecidos. No entanto, sua longa história faz com que muitos médicos ainda o utilizem, embora nesses casos seja desejável combiná-lo com um biomarcador mais sensível. O uso prévio de injeções intramusculares deve sempre ser excluído.
As transaminases e desidrogenase láctica e suas isoenzimas não devem ser utilizadas para o diagnóstico de infarto agudo, pois seu aumento pode ocorrer em outras circunstâncias clínicas: embolia pulmonar, acidose, lesões hepáticas e musculares, bem como pela administração de alguns medicamentos, como estatinas. Eles só serão usados quando for impossível ter marcadores mais específicos.
Assim, em uma síndrome coronariana aguda sem supradesnivelamento do segmento ST, a presença de marcadores bioquímicos anormais nos informará sobre a existência de angina instável, enquanto uma elevação deles confirmará que estamos diante de um infarto não Q.
Ecocardiograma
A ecocardiografia, na fase aguda dos eventos coronarianos, descobre anormalidades na motilidade das paredes em 89% a 100% dos pacientes com infarto transmural, com diminuição da sensibilidade entre 79% e 86% 6 para os não infartos. trasmural. Infartos pequenos podem não produzir anormalidades ecocardiográficas. Existem limitações técnicas devido às más janelas acústicas e à presença de entidades como bloqueios de condução e miocardite.
Previsão
Os pacientes podem ser classificados em 3 grupos de risco:
a) Alto risco . Aqueles com angina de repouso, recorrente ou acelerada e que também apresentam uma das seguintes características: instabilidade hemodinâmica (hipotensão, insuficiência cardíaca, disfunção mitral); Descida do ST> 1 mm transitória ou persistente; novo bloqueio de ramo ou arritmias ventriculares; angina pós-infarto (no primeiro mês); após revascularização coronariana (no primeiro mês); idade> 75 anos; troponinas marcadamente elevadas (troponina T> 0,1 ng / ml). Esses tipos de pacientes têm risco maior que 5% de sofrer infarto do miocárdio ou morrer nos primeiros 30 dias, portanto, o cateterismo cardíaco é recomendado nas próximas 24 horas para revascularização.
b) Risco médio . Pacientes com angina instável mais ausência de características de alto risco, inversão da onda T> 2 mm em 2 ou mais derivações e presença de algum modificador de risco (doença arterial coronariana prévia, doença arterial periférica conhecida ou doença cerebrovascular, diabetes mellitus, revascularização anterior, troponinas> 0,01 ng / ml).
c) Baixo risco . O restante dos pacientes sem características de alto ou médio risco e eletrocardiograma normal.
Complicações de ataque cardíaco
As complicações do infarto agudo do miocárdio são muito comuns. Eles podem aparecer no momento de desencadear o episódio ou desenvolver-se posteriormente, mas é nos primeiros dias que ocorre com mais frequência.
Essas complicações são: 1) arritmias e distúrbios da condução atrioventricular; 2) insuficiência cardíaca; 3) soc cardiogênico; 4) regurgitação mitral aguda; 5) defeito do septo ventricular agudo; 6) ruptura miocárdica; 7) pericardite e síndrome pós-infarto; 8) angina pós-infarto; 9) infarto do ventrículo direito; 10) embolia pulmonar e 11) embolia sistemática.
1. Arritmias . A incidência de arritmias no curso de um infarto agudo do miocárdio varia de acordo com os diferentes autores entre 70 e 100% dos casos. Isso depende de muitos fatores (idade, uso de drogas, acidose, tamanho do infarto, etc.), mas é fundamentalmente dado pelos diferentes métodos de detecção, pois quando se usa a monitoração por osciloscópio, sua frequência de aparecimento é menor do que quando é usado . faz com registro em memória eletrônica ou fita magnética. Seguindo Lown, é útil dividir as arritmias em dois tipos: 1) aquelas produzidas como resultado do processo isquêmico (falha elétrica), e 2) aquelas secundárias a uma descompensação do estado hemodinâmico devido à deterioração miocárdica grave (falha mecânica) .
Em última análise, ambos são consequência de uma alteração eletrofisiológica das miofibrilas, mas, nesta última, a terapia da falha da bomba também deve ser abordada.
As arritmias serão descritas resumidamente com base em seu local autônomo de origem.
a) Taquicardia sinusal. Denota hiperatividade simpática e ocorre em 30% dos casos; é mais frequente nos enfartes anteriores do que nos inferiores. As causas mais comuns são: dor, angústia, insuficiência cardíaca, pericardite e hipovolemia. As que requerem um diagnóstico diferencial preciso são a falha de bomba e a hipovolemia, pois o tratamento é muito diferente em ambas. Uma informação muito útil é o registro da pressão capilar pulmonar com um cateter de Swan-Ganz: sua medição decidirá se há hipertensão venocapilar (mais de 18 mm Hg) ou, ao contrário, hipotensão (menos de 19 mm Hg. )
b) Bradicardia sinusal. No momento inicial do infarto, ocorre em 40% dos casos, mas na unidade coronariana é observada em apenas 20%. É mais frequente em infartos menores do que em anteriores, na proporção de 3: 1, possivelmente devido à hiperatividade parassimpática.
É uma arritmia por instabilidade elétrica, por isquemia do nó sinusal, reflexos vagais ou pericardite. Às vezes, está associada à extrassístole ventricular devido ao aumento do automatismo
c) Extrassístole atrial. Ocorre em 20% dos casos e é devido a um aumento na automaticidade ou fenômenos de reentrada atrial, secundária a falha de bomba, hipóxia ou distúrbios eletrolíticos.
d) Fibrilação atrial. É observada em 10 a 15% dos pacientes e pode indicar insuficiência ventricular esquerda precoce ou infarto atrial. Também pode responder a causas extracardíacas, como hipocalemia, hipertireoidismo, hipóxia ou embolia pulmonar. Como a sístole atrial contribui com 15% da fração de ejeção ventricular, sua perda em um paciente com infarto pode ser essencial para a manutenção de um débito cardíaco adequado. Da mesma forma, se a resposta ventricular for elevada (maior que 130 batimentos / minuto), o estado hemodinâmico se deteriorará, observando-se sinais de descompensação cardíaca (oligúria, estertores basais, hipotensão) devido ao encurtamento do período de enchimento diastólico e aumento do miocárdio consumo de oxigenio. que por sua vez pode espalhar necrose.
e) flutter atrial. Presente em 1% a 2% dos IAM, como a fibrilação, pode ser decorrente de insuficiência ventricular esquerda ou infarto atrial. Dependendo do grau de bloqueio AV, a freqüência cardíaca variará de 75 a 300 batimentos / minuto. Em infartos inferiores, bloqueios 3: 1 e 4: 1 são geralmente observados devido a isquemia ou lesão do nó AV. Com esses graus de bloqueio, as ondas agitadas típicas são bem evidenciadas.
f) Taquicardia atrial paroxística. Ocorre em 5% dos pacientes com infarto. A freqüência cardíaca varia entre 150 e 300 batimentos por minuto e sua fisiopatologia é a mesma da extra-sístole atrial.
g) Ritmo da junção AV (nodal). Sua frequência varia entre 2% e 15% de acordo com diferentes autores. Constitui ritmo de escape, devido à depressão do nó sinusal ou bloqueio AV em consequência de isquemia.
h) Bloqueio atrioventricular (bloqueio AV). O bloqueio AV de primeiro grau ocorre em 9% dos casos, segundo grau em 8% e terceiro grau em 7% dos pacientes internados na Unidade Coronária com IAM.
De todo o sistema de condução atrioventricular, a junção AV é o setor mais sensível. anoxia e isquemia. Por esse motivo, nos infartos agudos inferiores é comum observar diferentes graus de distúrbios da condução AV em decorrência do comprometimento do suprimento sanguíneo normal do nó AV.
O bloqueio AV de primeiro grau (todos os estímulos cruzam a junção AV, mas o fazem mais lentamente), ou tempo de condução AV de mais de 0,20 segundos, não requer tratamento, mas requer controle cuidadoso, pois pode evoluir para bloqueio de graus mais elevados; o último é visto com mais frequência em ataques cardíacos anteriores.
Os bloqueios AV de segundo grau (nem todos os estímulos cruzam a junção) respondem a dois mecanismos básicos. No Mobitz tipo I (ou bloqueios com períodos de Wenckebach) observa-se um prolongamento progressivo do segmento PR, o que indica uma condução cada vez mais afetada, até que uma onda P seja totalmente bloqueada. Isso prolonga o tempo disponível para o nodo AV se recuperar, e a onda P seguinte apresentará condução AV normal, o que dá origem a um novo ciclo de prolongamento progressivo do PR.
Esse tipo de bloqueio AV é mais frequente em infartos inferiores e geralmente se deve à isquemia do nó AV. Mesmo sem tratamento pode desaparecer em 72 horas. O bloqueio AV do tipo Mobitz I constitui 90% dos bloqueios AV de segundo grau.
O bloqueio AV de segundo grau do tipo Mobitz II é menos comum e é observado em menos de 10% de todos os pacientes com IAM que apresentam um bloqueio. Mostra a falta de condução abrupta de uma onda P sem alongamento prévio do segmento PR. Na maioria dos casos, está associado a um infarto anterior e quase sempre se deve a uma lesão abaixo do feixe de His, que freqüentemente evolui para bloqueio completo. Portanto, esse distúrbio requer, em geral, o implante de marca-passo provisório no ventrículo direito.
No bloqueio AV de terceiro grau (completo), os átrios e os ventrículos batem independentemente.
É observada em pacientes com infartos inferiores ou anteriores, e se origina no edema que envolve o tecido infartado e compromete o sistema de condução. Nos infartos inferiores a lesão é nodal, com desenvolvimento gradual do bloqueio e aparecimento de ritmos da junção AV de 40 a 60 batimentos por minuto. Em infartos anteriores, o bloqueio AV se instala abruptamente e é uma causa de alta mortalidade. É causada por extensa necrose do septo envolvendo o feixe de His (lesão infranodal) e pode terminar repentinamente em assistolia, falha de bomba ou fibrilação ventricular. O bloqueio AV de terceiro grau é uma indicação formal para um marca-passo endocavitário temporário e estimulação sob demanda.
i) Distúrbios da condução intraventricular. Eles são vistos em 10 a 20% dos casos. Eles podem ser: 1) bloqueio completo de ramo direito (RBBB); 2) bloqueio completo do ramo esquerdo (BRE); 3) hemibloqueio anterior esquerdo (HAJ); 4) hemibloqueio posterior esquerdo (HPI); 5) BCRD com HAI e 6) BCRD com HPI.
Bloqueios de ramos completos podem progredir para bloqueio AV de terceiro grau no curso de um infarto agudo. IAM complicado com RBBB ou RBBB têm alta mortalidade hospitalar.
j) Extrassístole ventricular. É a arritmia mais comumente observada no IAM, com frequência superior a 75% dos casos. É devido a um desequilíbrio elétrico ao nível celular produzido pela isquemia (arritmia por instabilidade elétrica). Como em geral não há descompensação hemodinâmica, não são de mau prognóstico, mas devem ser tratados se aparecerem a mais de seis por minuto, se forem multifocais, bigeminados ou trigeminados ou se já causaram arritmias graves anteriormente.
k) Taquicardia ventricular. Ocorre em 20% dos pacientes nas primeiras horas do infarto agudo. Geralmente é definida como uma sucessão de batimentos ventriculares (quatro ou mais) com uma frequência inferior a 120 batimentos / minuto.
É mais frequente nos casos de infarto transmural e naqueles com insuficiência ventricular grave, e o mecanismo eletrofisiológico responde ao aumento do automatismo das fibras de Purkinje lesadas (foco ectópico) e aos fenômenos de reentrada. É uma arritmia grave que requer decisões de tratamento rápidas.
l) Ritmo idioventricular acelerado. É definido como um ritmo ventricular com uma frequência de 60 a 100 batimentos por minuto e ocorre em 15% dos pacientes com IAM. É observada no segundo ou terceiro dia do evento e responde a um mecanismo de escape, devido à desaceleração do ritmo de base ou a um foco ectópico de taquicardia ventricular. Os episódios costumam ser temporários e de bom prognóstico.
m) Fibrilação ventricular. Essa arritmia grave se manifesta no ECG por uma sucessão de ondas lentas, polimórficas e de início irregular. Ocorre em 10% dos casos e é a principal causa de morte em pacientes com IAM antes de sua entrada na Unidade Coronariana. Pode ser elétrico ou primário e mecânico ou secundário. O primeiro é o que ocorre como um evento abrupto em um paciente sem evidência prévia de insuficiência circulatória, enquanto o mecânico é a arritmia terminal em um paciente com grave falha de bomba.
2. Insuficiência cardíaca (falha da bomba) . A insuficiência ventricular esquerda no curso de um IAM continua sendo o desafio mais importante para o médico da Unidade Coronariana. A perda do miocárdio funcionante é a causa mais importante de insuficiência cardíaca nessa condição, mas em muitos casos existem outros fatores que contribuem para o desencadeamento, manutenção ou agravamento da insuficiência ventricular. Estes incluem a diminuição da pré-carga devido à hipovolemia relativa ou absoluta, o aumento da pós-carga causado pela descarga adrenérgica nas primeiras horas da I AM, a presença de arritmias e complicações mecânicas pós-infarto, como discinesia ventricular. , ou ruptura do músculo papilar ou do septo interventricular.
Do ponto de vista clínico, seguindo os critérios de Killip e Kimball, os pacientes são classificados em quatro classes funcionais:
Classe I: pacientes sem complicações, sem manifestações de insuficiência cardíaca.
Classe II: sinais ou sintomas mínimos de insuficiência cardíaca (estertores basais, galope ou hipertensão venosa).
Classe III: edema agudo de pulmão devido a grave
insuficiência ventricular esquerda.
Classe IV: choque cardiogênico.
A monitorização hemodinâmica em Unidade Coronária, com a colocação de cateter de termodiluição de Swan-Ganz no leito capilar da artéria pulmonar (que representa as pressões diastólicas finais do ventrículo esquerdo), é de grande importância para uma avaliação correta desses pacientes. Considerando o estado de congestão pulmonar e perfusão periférica, Forrester e Ganz os classificam como:
Grupo I: índice cardíaco maior que 2,2 1 / min / m2 e pressão capilar pulmonar menor que 18 mm Hg. O paciente é assintomático.
Grupo II: pressão capilar pulmonar superior a 18 mm Hg. e o débito cardíaco permanece acima de 2,2 1 / min / m2.
O paciente pode ser assintomático ou apresentar sinais clínicos e radiológicos de congestão pulmonar mais ou menos intensa. Se o quadro clínico for de edema agudo de pulmão e a resistência sistêmica for muito elevada, também pode apresentar sinais de vasoconstrição periférica que simulam uma situação de baixo débito.
Grupo III: pressão capilar pulmonar inferior a 18 mm Hg. e débito cardíaco menor que 2,2 1 / min / m2. Nesta situação, que é hipovolemia absoluta (devido a vômitos, diarreia ou diurese excessiva) ou relativa (em que não há hipovolemia verdadeira, mas devido à contratilidade diminuída, o ventrículo precisa de uma pré-carga maior para manter a função normal), o paciente pode ser assintomático ou apresentar sinais de hipoperfusão periférica e hipotensão arterial.
Grupo IV: pressão capilar pulmonar maior que 18 mm Hg. e o índice cardíaco menor que 2,21 / min / m2. Nessa situação de grave deterioração hemodinâmica, o paciente praticamente sempre apresenta sinais clínicos de congestão pulmonar e baixo débito. A pressão arterial geralmente está baixa. A forma mais séria de insuficiência cardíaca, o choque cardiogênico, está associada a esse grupo. Para fazer o diagnóstico de choque cardiogênico, é necessário que a pressão capilar pulmonar esteja elevada e, assim, descartar choque hipovolêmico.
3. Choque cardiogênico . La Myocardial Infarc¬tion Research Unit (MlRU) do National Heart and Lung Institute de Bethesda define choques cardiogênicos asi al:
a) Pressão arterial sistólica menor que 90 mm Hg ou menor que 30 mm Hg. ao seu valor anterior em um hipertenso, por pelo menos meia hora, associado a:
b) Perfusão periférica inadequada manifestada pelos seguintes critérios bioquímicos clínicos:
1. Débito urinário menor que 20 ml / hora e natriúria menor que 30 mEq / 1.
2. Pele fria, pálida, lívida, suada e viscosa.
3. Desordem mental (hipoperfusão cerebral) caracterizada por agitação, sonolência ou coma.
4. Compromisso do metabolismo celular aeróbio (acidose láctica).
A incidência de choque cardiogênico após IAM é de aproximadamente 15% e a mortalidade é avassaladora, ultrapassando 90% em algumas séries. Estudos anatomopatológicos mostraram que essa condição ocorre quando mais de 40% da massa miocárdica está lesada, seja por lesão recente ou associada a necrose antiga. A extensão do dano está diretamente relacionada ao prognóstico e é resultado de uma lesão progressiva, constituindo um ciclo vicioso de isquemia, depressão miocárdica e aumento da isquemia.
A necrose extensa da massa ventricular causa redução da contratilidade, diminuição do débito cardíaco e diminuição da pressão arterial. O volume sistólico cai rapidamente e, se essa queda atingir entre 30 e 50% do seu valor normal, ocorre hipoxemia tecidual, com conseqüente metabolismo celular anaeróbio e desenvolvimento de acidose láctica.
Por meio da hipoxemia e da liberação de catecolaminas (mediadas pela ativação simpática), um mecanismo de feedback negativo é instalado com um aumento consecutivo da resistência vascular, principalmente no rim, no território esplâncnico e na pele, de forma que o volume de sangue o retorno ao coração direito diminui.
Isso explica que, embora a pressão diastólica final do ventrículo esquerdo aumente levemente, em 75% dos casos não há edema pulmonar ou aumento da pressão venosa central.
Hipotensão, liberação de noradrenalina e redução do volume circulante efetivo levam à hipoperfusão renal e hepática. O primeiro estimula o sistema renina-angiotensina-aldosterona e a redução da perfusão hepática reduz o metabolismo da aldosterona no fígado. Esses dois mecanismos produzem aumento da aldosteronemia, com conseqüente retenção de sódio e perda de potássio em nível renal. Necrose miocárdica, anoxia, acidose e deficiência de potássio são terreno fértil para o desenvolvimento de arritmias graves.
Manter baixo débito cardíaco e hipoperfusão por várias horas pode causar maior dano miocárdico, estabelecendo um feedback positivo e irreversível entre diminuição do débito cardíaco e dano miocárdico. O mecanismo desse círculo vicioso parece responder à hipoperfusão miocárdica e a um fator humoral que deprime a contratilidade das células miocárdicas. A hipoperfusão do miocárdio, com a conseqüente diminuição da contratilidade, estende a área infartada. Dentre os fatores humorais, destaca-se a produção de polipeptídeos ativos que deprimem a força de contração miocárdica e que são produzidos principalmente no pâncreas e no território esplâncnico devido à ruptura de lisossomas devido à hipóxia desses tecidos.
Assim, o ciclo vicioso que surge das complicações do choque, se não resolvido precocemente, leva à morte rapidamente. Em relação às características cômicas desta grave condição, pode-se acrescentar que os pacientes estão gravemente comprometidos, com sintomas e sinais secundários à má perfusão tecidual, tais como: confusão mental, inquietação, vasoconstrição de tegumentos, cianose periférica, pulsos estreitos, hipotensão, taquicardia , oligúria (diurese inferior a 20 ml / hora), etc., e congestão pulmonar: dispneia, ortopneia, polipneia, etc. Os indicadores mais sensíveis de perfusão tecidual são o estado do sistema nervoso central e o fluxo urinário, elementos que devem ser avaliados permanentemente.
Para se chegar ao diagnóstico de choque cardiogênico por IAM, os seguintes elementos devem ser considerados:
A) dor torácica prolongada nas últimas 48 horas;
B) às vezes edema agudo de pulmão sem doença valvar cardíaca prévia, uremia ou intoxicação;
C) choque não decorrente de intoxicação, sepse ou hemorragia;
D) angor rebelde.
Qualquer um dos casos acima também deve apresentar sinais eletrocardiográficos de infarto agudo e aumento das enzimas do parasita.
Um paciente que retina com os critérios acima, e também aqueles mencionados na definição original, está em choque cardiogênico.
Para quantificar os distúrbios hemodinâmicos, sua evolução e o efeito das medidas terapêuticas, deve-se fazer um controle com aparelhos de monitorização hemodinâmica. Um instrumento muito útil e já citado é o cateter de Swan-Ganz, que possibilita medir o débito cardíaco, as pressões no circuito pulmonar e o conteúdo de 02 do sangue venoso misto.
4. Regurgitação mitral aguda . Pode ser devido a disfunção ou ruptura do músculo papilar. A incidência de ruptura de um músculo papilar e / ou de suas cordas tendíneas no infarto agudo é de 1%. Ocorre com mais frequência em infartos inferiores e é clinicamente caracterizada por um sopro pansistólico de regurgitação mitral com irradiação para a axila, dorso e borda esquerda do esterno. O quadro clínico pode variar de insuficiência cardíaca esquerda leve a edema pulmonar agudo.
Cateterismo direito com Swan-Ganz mostra a existência de ondas "V" proeminentes na monitoração da pressão venocapilar pulmonar, por transmissão retrógrada. O conteúdo de oxigênio do átrio e ventrículo direitos é igual, o que faz o diagnóstico diferencial com CIV. Nesse sentido, o ecocardiograma bidimensional e, melhor ainda, o ecodoppler são muito úteis para esclarecer os quadros duvidosos. Embora o ecocardiograma bidimensional possa às vezes não revelar uma ruptura do músculo papilar ou septal, o Doppler pode demonstrar a anormalidade de fluxo resultante. Ressalta-se que a sobrevida média em pacientes com ruptura parcial de músculo papilar é de três dias, sendo necessário diagnóstico imediato para correção cirúrgica.
5. Comunicação ventricular (VSD) em AMI . A ruptura do septo interventricular ocorre em menos de 1% dos pacientes com IAM ântero-septal ou póstero-septal. A maioria dos casos ocorre na primeira semana do episódio agudo. O tamanho do defeito e a presença ou ausência de aneurisma septal determinam as consequências hemodinâmicas. É uma complicação que leva à mortalidade: mais de 25% dos casos morrem nos primeiros três dias, 65% nos primeiros quinze dias e 90% nos primeiros dois meses.
Os achados clínicos incluem um froito sistólico palpável e um sopro holossistólico rude no quarto ou quinto espaço intercostal esquerdo, irradiando a borda esternal esquerda, o ápice e a axila. A insuficiência cardíaca é global. Às vezes, a diferença auscultatória entre um sopro de VSD e aquele de regurgitação mitral aguda pode ser difícil. Um cateter de Swan-Ganz deve ser colocado e amostras de sangue retiradas da veia cava superior, do átrio direito, do ventrículo direito e da artéria pulmonar, para determinar o conteúdo de oxigênio do mesmo. Se a diferença entre o átrio direito e o ventrículo direito for de 1 ou mais volumes, o diagnóstico de CIV é feito, uma vez que é mostrado um shunt da esquerda para a direita.A ecocardiografia bidimensional também pode ser usada para detectar e localizar o defeito septal pós-infarto. Se o VSD for pequeno, pode não ser visualizado por eco, mas por eco-Doppler. Se o ecocardiograma bidimensional não encontrar um CIV e o Doppler não detectar fluxo turbulento na região do septo interventricular, a causa do sopro (e dos sintomas) será quase invariavelmente regurgitação mitral aguda.
6. Ruptura cardíaca externa . Essa complicação é responsável por cerca de 10% dos casos fatais intra-hospitalares. É quatro vezes mais comum em mulheres, especialmente em pacientes com mais de 60 anos que são hipertensos. Geralmente ocorre na primeira semana (terceiro ao quinto dia) e é observada principalmente em infartos anteriores transmurais que ocupam pelo menos 10% da circunferência ventricular esquerda. Clinicamente, é caracterizada por reaparecimento súbito de dor torácica, colapso hemodinâmico abrupto, hipotensão com sinais claros de tamponamento cardíaco e dissociação eletromecânica. Nessa situação, enquanto as manobras de ressuscitação estão sendo iniciadas, deve-se tentar a pericardiocentese.Se um ecocardiograma for bem-sucedido assim que a ressuscitação começar, a presença ou ausência de hemopericárdio pode ser rapidamente confirmada.
7. Pericardite . A pericardite epistenocárdica é muito comum (15% dos casos), surgindo entre o primeiro e o segundo dia e desaparecendo após uma semana. É caracterizada por dor torácica persistente que pode se intensificar com movimentos respiratórios ou alterações de decúbito, com irradiação para os ombros e costas. A presença de esfregaço sistodiastóico confirma o diagnóstico.
O aparecimento de atrito pleural é comum e a existência de episódios de pericardite recorrente associada à pneumonite pode ser a expressão da síndrome autoimune de Dressier ou síndrome pós-infarto.
8. Angina pós-infarto . Aproximadamente 10 a 15% dos pacientes com infarto agudo desenvolverão angina instável na primeira semana do evento. Seu prognóstico é grave, pois é uma complicação que está associada a uma alta incidência de infarto recorrente ou óbito durante a internação. Além disso, a mortalidade desses pacientes é alta 3 a 6 meses após o evento.
Este quadro de angina recorrente, ou angina pós-infarto precoce, pode ser devido a: 1) aumento transitório na demanda de oxigênio, taquiarritmias, anemia, drogas, alterações no volume sanguíneo ou problemas cardíacos mecânicos, como aneurisma ventricular, regurgitação mitral, VSD ou insuficiência cardíaca grave; 2) queda transitória no suprimento de oxigênio ao miocárdio, que pode responder à trombose transitória ou agregação plaquetária em artérias gravemente danificadas ou a um espasmo da artéria coronária próximo ao local de uma estenose aterosclerótica.
Considerando as múltiplas causas possíveis, se o paciente tem angina recorrente, deve-se investigar por que a angina é recorrente. Uma avaliação clínica completa, testes laboratoriais e até mesmo uma angiografia coronariana devem ser realizados se for esperado que o paciente se beneficie da terapia invasiva.
Mesmo a angina mais recente, mas que ocorre dentro de um mês do IAM e com o paciente ainda em repouso, costuma responder por causas semelhantes e seu prognóstico é grave, para o qual é necessário seguir as orientações acima citadas.
9. Infarto do ventrículo direito (AMI-RV). Embora o IAM isolado do ventrículo direito seja muito raro, essa condição pode estar associada a um infarto transmural da parede posteroinferior do ventrículo esquerdo.
A síndrome clínica do IAM-VD aparece entre 3% e 8% dos infartos póstero-inferiores, embora seja encontrada entre 15 e 34% das autópsias e em 30-70% dos infartos inferiores estudados com cintilografia e ecocardiografia. Essas diferenças se devem ao fato de que muitas das lesões patológicas não apresentam tradução clínica ou hemodinâmica. O espectro clínico que lhe dá origem é muito amplo - pode até levar ao choque cardiogênico - mas seu diagnóstico se baseia em evidências clínicas e hemodinâmicas de disfunção cardíaca com preponderância certa, no curso de IAM. eletrocardiográfico de infarto agudo posteroinferior, corroborado por elevação enzimática; 2) evidência clínica de insuficiência cardíaca direita dominante, demonstrada por ingurgitamento jugular,hipertensão venosa central maior que 18 cm H20; e 3) confirmação hemodinâmica com cateter de Swan-Ganz de elevação da pressão da diástole final do ventrículo direito ou valores iguais ou maiores que a pressão capilar pulmonar. Também é acompanhada por distúrbios da condução AV em um alto percentual de casos, que em algumas séries chega a 100%.
A presença de insuficiência direita no curso de um IAM também pode responder à insuficiência ventricular esquerda ou tromboembolismo pulmonar. Corresponde a estabelecer o diagnóstico diferencial com essas entidades.
10. Embolia pulmonar . Pode causar morte súbita, arritmias e / ou insuficiência cardíaca refratária no curso de um IAM. Sua frequência de aparecimento não é conhecida. Na maioria dos casos, o ponto de partida são as veias profundas dos membros inferiores e da pelve.
O quadro clínico dependerá do número e do tamanho dos êmbolos. Assim, pode-se observar desde sudorese, dor torácica, dispneia e hipotensão moderada, até insuficiência cardíaca refratária ao tratamento, arritmias, choque e morte. A ausculta revelará apenas "um segundo ruído duplo e fixo, com P2 acentuado. A arritmia mais frequente é a fibrilação atrial, e o ECG mostrará desvio do eixo para a direita, rotação no sentido horário, aumento atrial direito e bloqueio completo do ramo direito .Para confirmar o diagnóstico, o estudo mais útil é a cintilografia radioisotópica pulmonar. Se mostrar grandes áreas de hipoperfusão bilateral, uma arteriografia pulmonar deve ser indicada para determinar a necessidade de uma embolectomia.
11. Embolias sistêmicas . É uma complicação rara, devida à formação de trombos murais na parede do ventrículo esquerdo ou átrio infartado, de onde migram para a periferia. Seu prognóstico depende do órgão impactado (rim, cérebro, membros inferiores, etc.) e do tamanho e hierarquia da artéria ocluída.