
 Português
Português  Español
Español  English
English Carlos J. Galli Mainini
Síndrome de Adams-Stokes . veja síndrome de Stokes-Adams
Síndrome de Adie . Síndrome neurológica de etiologia desconhecida. É caracterizada por uma reação patológica da pupila à acomodação (miotonia), na qual a pupila do lado afetado se contrai e dilata mais lentamente do que no lado saudável. Certos reflexos do tendão estão ausentes ou diminuídos, mas não há outros distúrbios motores ou sensoriais ou sinais de doença do sistema nervoso central.
Síndrome adiposogenital de Frohlich . Síndrome endocrinogenital devido à disfunção hipotalâmica. Suas características básicas são: a) obesidade feminoide com hipoplasia das gônadas: e ausência de características sexuais secundárias; b) diabetes insipidus; ec) retardo mental e de crescimento e distúrbios da visão. É produzida por tumores ou outros processos patológicos que afetam a região hipotalâmica da hipófise e que incluem o centro hipotalâmico do apetite, responsável pela obesidade.
Síndrome adrenogenital . Conjunto de quadros clínicos produzidos por hiperfunção adrenal e cujas manifestações são predominantemente genitais. Inclui o seguinte: a) síndrome de hiperplasia adrenal congênita. causada por deficiência específica de enzimas, que estão envolvidas no metabolismo dos corticosteroides (ver Cap. 61); b) a síndrome de virilização adrenal em adultos, produzida por tumores adrenais benignos ou malignos; ec) a síndrome de feminização adrenal, causada por tumores ou por deficiência enzimática.
Síndrome da afasia motora . Síndrome neurológica devido à oclusão da divisão superior da artéria cerebral média. O quadro clássico é caracterizado por: a) o indivíduo retém sua linguagem interna (sabe o que dizer), mas perdeu a memória dos movimentos necessários para fazê-lo, de modo que não consegue expressar essa linguagem em palavras: articulada (anartria); b) compreende o que está sendo dito, e a leitura e a escrita são preservadas; c) está totalmente ciente do que está acontecendo, o que muitas vezes leva à exasperação, ed) em alguns casos, distúrbios variáveis de compreensão estão associados. É devido a lesões nas regiões frontal inferior, parietal anterior e insular anterior, de origem embólica e às vezes devido a tumores cerebrais ou abscessos.
Síndrome de afasia sensorial . Síndrome neurológica devido à oclusão da artéria cerebral média esquerda. Resulta da perda da capacidade de compreensão do significado das palavras (afasia de compreensão), sem alterações na articulação das palavras. O indivíduo é privado de todos os meios de comunicação social. Há: a) incapacidade do paciente de se fazer compreender (nem verbalmente nem por escrito), porque as palavras corretas da língua são substituídas por outras de som ou significado mais ou menos semelhante (parafasias literais e verbais), ou por neologismos ou jargão incompreensível; b) incapacidade de encontrar o nome dos objetos que percebe;c) incapacidade de compreender o que lê ou o que lhe é dito ed) sentir, por parte do observador, que o paciente não tem uma consciência clara do que lhe está acontecendo.
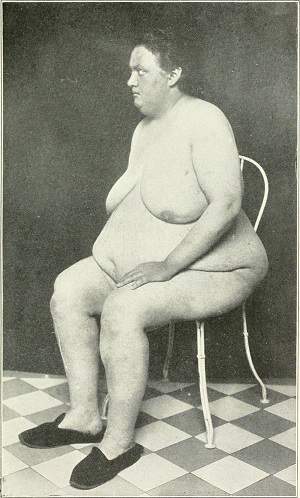 Síndrome de Frohlich 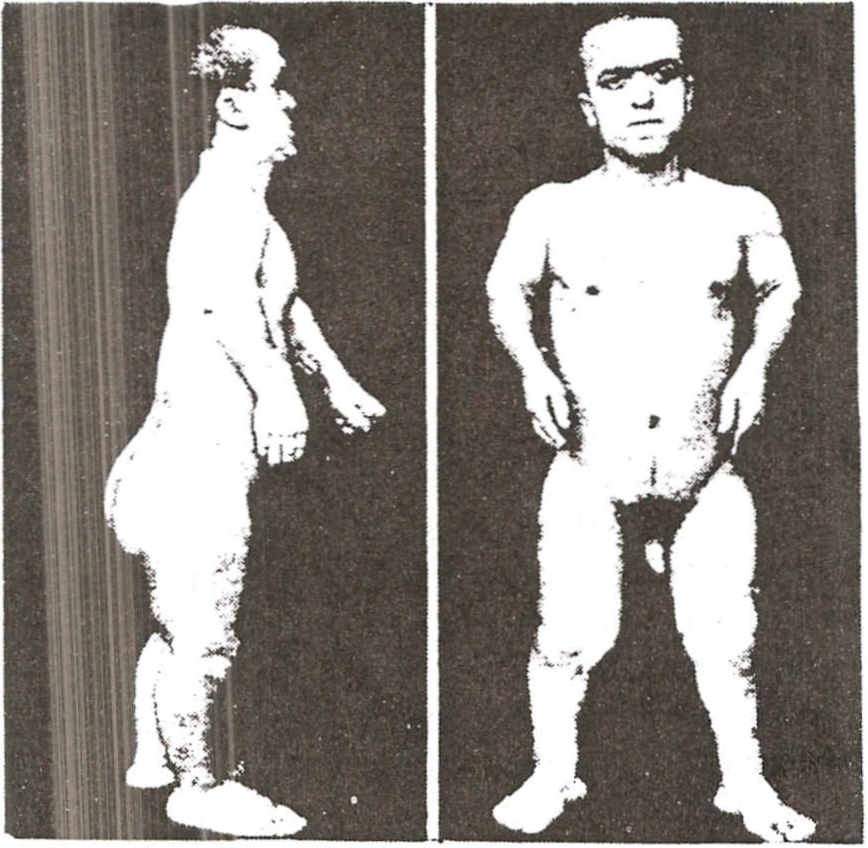 Síndrome de acondroplasia (Suros, Medical Semiology, 1968) |
Síndrome de agnosia visual . Síndrome neuro-oftalmológica de origem central. É produzida por lesões que afetam as vias visuais e as áreas corticais primária, secundária e terciária da visão, e a imagem é de um indivíduo que vê bem os objetos, mas não consegue reconhecê-los a menos que, conforme o caso, toque, cheire ou saboreá-los, ou reconhecer os sons que eles fazem.
Síndrome do buraco óptico . Síndrome neurológica da base do crânio. Cegueira unilateral, de início insidioso e de caráter progressivo, acompanhada ou não de dor ipsilateral com irradiação para o pescoço e região mandibular. É causada, quase sempre, por um glioma do nervo óptico.
Síndrome do buraco anterior irregular . veja a síndrome de Bonnet
Síndrome do buraco irregular posterior . veja Síndrome de Vernet
Síndrome de Ahumada-Del Castillo . veja síndrome de Forbes-Albright
Síndrome menor da asa do esfenóide . veja a síndrome de Kennedy
Síndrome de Albright-McCune-Sternberg . Síndrome hereditária caracterizada por distúrbios ósseos, cutâneos e endócrino-ginecológicos. É uma condição rara observada em meninas e se manifesta por displasia óssea (espessamento, curvatura e fraturas fáceis dos ossos), anormalidades pigmentares da pele (manchas com bordas irregulares, cor de café com leite) e puberdade precoce com início desenvolvimento de características sexuais secundárias.
Síndrome do álcool fetal . Síndrome do bebê devido ao uso abusivo de álcool pela mãe durante a gravidez. As seguintes características podem estar presentes, isoladas ou em combinação: a) baixa estatura em relação ao que corresponderia em função do peso; b) luxação facial (hipoplasia da mandíbula, fenda palatina) e ocular (microftalmia, prega epicanto) e luxação bilateral do quadril; c) anomalias cardíacas e genitais ed) malformações cardíacas e renais. Além disso, o recém-nascido pode desenvolver um quadro semelhante; apenas que é persistente, o da abstinência do álcool em adultos, e com o tempo mostra um retardo mental de grau variável.
Síndrome de Alexia sem agrafia . Síndrome neurológica devido a lesão do lobo occipital. Hemianopia direita e incapacidade de ler em voz alta e compreender a linguagem escrita, sem alterações no ditado ou leitura espontânea no silêncio. É devido a uma lesão do lobo occipital esquerdo, geralmente tumoral, associada a uma lesão do corpo caloso.
 Síndrome de Albright-McCune-Sternberg Síndrome de Albright-McCune-Sternberg
|
Síndrome de Andersen . Síndrome de mucoviscidose complicada. É uma tríade de mucoviscidose (fibrose cística do pâncreas), bronquiectasia e hipovitaminose A, em que o primeiro componente é a doença de base e os outros dois são complicações frequentes.
Síndrome do ângulo cerebelopontino . Síndrome neurológica central com envolvimento dos nervos cranianos V, VII e VIII. É manifestada por surdez perceptual (NC VIII), seguida de arreflexia labiríntica ou hiporreflexia (NC VIII), e às vezes associada à arreflexia corneana (NC V) e paralisia facial de intensidade variável (NC VII). É causada por meningiomas ou neurinomas do nervo acústico.
Síndrome de ansiedade . Síndrome funcional dos estados de ansiedade. Palidez, sudorese, tremor variável, palpitações, respiração rápida e superficial ou respiração "curta", medo injustificado, etc., sem causa orgânica detectável. É observada em indivíduos submetidos a situações sustentadas de tensão psíquica.
Síndrome de esmagamento . Síndrome por esmagamento ' realizada em qualquer setor do corpo. Ele define o quadro de edema, oligúria, hipovolemia, choque e, finalmente, insuficiência renal aguda, que é freqüentemente observada em indivíduos que sofreram lesões por compressão traumáticas prolongadas, principalmente quando o trauma afetou uma massa considerável de tecido muscular (pág., Esmagamento de ambos os membros inferiores como resultado de um colapso).
Síndrome da apnéia do sono . veja Síndrome de hipersonia com apnéia noturna
Síndrome da artéria mesentérica superior . Síndrome abdominal causada por obstrução extrínseca do trânsito duodenal. É causada pela artéria mesentérica superior por compressão da terceira porção do duodeno contra o tronco da aorta abdominal, em consequência da qual ocorre uma obstrução aguda e transitória, mas recorrente com o tempo, do segmento em questão. As manifestações da síndrome são altamente variáveis: desde distúrbios mínimos até náuseas e vômitos pós-prandiais, dor abdominal central e, ocasionalmente, distensão extrema do estômago e duodeno.
Síndrome de alça aferente . Síndrome secundária à gastrojejunostomia. É manifestada por episódios pós-prandiais recorrentes de náusea, distensão abdominal e dor na parte superior do abdome. A condição é atribuída à obstrução parcial crônica intermitente da alça intestinal proximal (duodeno e jejuno)
Síndrome de alça cega . Síndrome de disfunção do intestino delgado. É caracterizada, em suas formas graves, por esteatorreia, má absorção de vitamina B 12 e anemia não ferropriva. É atribuída à proliferação bacteriana anormal do intestino delgado, como consequência da estenose do órgão ou de operações que deixam as alças intestinais com pouco trânsito ativo.
Síndrome de Ashernan . Síndrome de amenorréia por destruição endometrial. Amenorréia secundária e esterilidade, com cicatrizes e aderências intrauterinas, e desaparecimento mais ou menos completo do endométrio. É causada por procedimentos instrumentais (curetagem), geralmente de hemorragia pós-parto ou aborto complicado por infecção.
Síndrome de dupla atetose . veja síndrome de Vogt .
Síndrome de Aubertin . Síndrome de interrupção abrupta do fluxo sanguíneo nas câmaras cardíacas. É um quadro de aparecimento súbito e rapidamente progressivo de dor retroesternal aguda, dispneia, taquipneia, angústia, lividez, pulso filiforme, colapso e quase sempre morte, que se observa como complicação da estenose mitral e ocorre quando um coágulo se desprende do corpo. o átrio esquerdo oclui abruptamente o forame atrioventricular.
Síndrome auriculotemporal . veja a síndrome de Frey .
Síndrome de Avellis . Síndrome neurológica do tronco encefálico com alterações motoras e sensoriais e envolvimento do nervo craniano X. Paralisia de corda vocal e palato mole no lado da lesão (X nervo craniano), e no lado oposto hemiplegia respeitando a face e perda da sensibilidade térmica e dolorosa (lesão do teto do bulbo e feixe piramidal). A causa é tumor ou amolecimento do cérebro.
Síndrome de Ayerza . Síndrome de insuficiência pulmonar crônica avançada Cianose grave , insuficiência cardíaca congestiva e vários graus de policitemia.
Síndrome de Babinski-Nageotteo . Síndrome neurológica da medula oblonga unilateral. Hemiplegia e hemianestesia no lado oposto da lesão e hemiasinergia, hemiataxia e lateropulsão no mesmo lado da lesão; e se as fibras simpáticas da formação reticular são afetadas, também miose, enoftalmia e ptose palpebral do lado da lesão. Geralmente é devido a uma obstrução vascular acima do cruzamento do feixe piramidal.
Síndrome de Babinski-Vaquez . Síndrome neurológica e vascular da sífilis avançada. É uma condição variável e polimórfica, que depende da localização e da gravidade das lesões principais e que se manifesta, basicamente, por alteração dos reflexos tendinosos (arreflexia patelar e aquiliana) e da pupila (arreflexia pupilar, miose, etc. .), anomalias. líquido cefalorraquidiano, estado de demência e regurgitação aórtica evidente.
Síndrome de Bafuerstedt . Síndrome de linfadenose cutânea benigna. Hiperplasia inflamatória dos folículos linfáticos da pele, afetando preferencialmente a face e as orelhas, em adultos jovens, na forma de nódulos solitários ou disseminados e que geralmente regridem espontaneamente.
Síndrome de Bandl-Frommel . Síndrome de ruptura iminente do útero. É típica da segunda etapa do trabalho de parto e se caracteriza pela tetanização do corpo uterino e do anel de Bandl, e pela possibilidade de palpar duas cordas duras e tensas que correspondem aos ligamentos redondos através da parede abdominal.
Síndrome de Banti . Síndrome da esplenomegalia congestiva crônica. É decorrente de processos que produzem hipertensão crônica do sistema porta, com hiperesplenismo secundário e é caracterizada por: a) esplenomegalia; b) anemia, leucopenia e trombocitopenia, com hiperplasia da medula óssea ec) cirrose hepática, com ascite e hemorragia por ruptura de varizes esofágicas.
Síndrome de Bard e Pic . Síndrome do carcinoma da cabeça do pâncreas. É manifestada por uma tríade característica: icterícia crônica progressiva, grande aumento da vesícula biliar e caquexia de evolução rápida. É devido à compressão do ducto biliar comum pela massa tumoral.
 Síndrome de Barraquer-Simmons Síndrome de Barraquer-Simmons 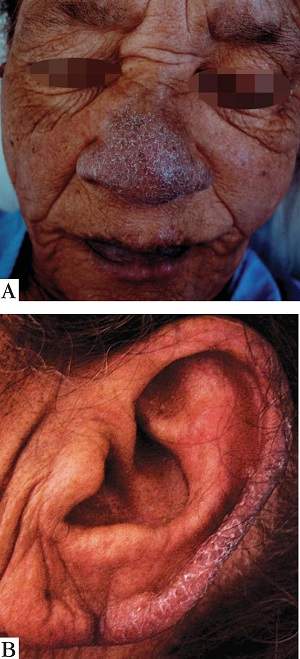 Síndrome de Bazex |
Síndrome de Barraquer-Simmons . Síndrome de lipodistrofia progressiva. Emagrecimento profundo da parte superior do corpo, especialmente o rosto, e grande obesidade na metade inferior, especialmente nas nádegas e coxas. É visto em mulheres jovens e a causa é desconhecida.
Síndrome de Barré-Lieou . Síndrome relacionada às condições orgânicas da coluna cervical. É caracterizada por cefaleia, distúrbios visuais temporários, fenômenos vasomotores na face, vertigem, zumbido, parestesia craniofacial, disfagia e disfonia intermitente, etc. É causada por irritação direta dos nervos espinhal e simpático, em pacientes com osteoartrite avançada do as três últimas, vértebras cervicais, ou por doenças vasculares (vertebrobasilar) ou psicogênicas.
Síndrome de Barrett . Síndrome de esofagite crônica. Inflamação crônica da mucosa do terço inferior do esôfago, associada ou não ao desenvolvimento de úlceras pépticas, que leva progressivamente à estenose por retração da cicatriz. A causa básica é, em geral, a substituição do epitélio escamoso ou escamoso do esôfago. por um epitélio secretor do tipo gástrico.
Síndrome de Bartter . Síndrome de hiperaldosteronismo secundário com hiperplasia de células justaglomerulares. É caracterizada por alcalose hipocalêmica e aumento acentuado dos níveis de renina plasmática, sem hipertensão arterial ou resposta à ação pressora da angiotensina. A causa é provavelmente genética e a condição pode ser acompanhada de retardo mental e baixa estatura.
Síndrome de Bassen-Kornzweig . Síndrome caracterizada por anormalidades da b- lipoproteína e outras alterações. Manifesta-se pela ausência de lip-lipoproteínas plasmáticas, retinite pigmentosa, acantocitose de hemácias e distúrbios neurológicos progressivos que começam na infância e se manifestam por ataxia, arrefexia e alterações graves na sensibilidade proprioceptiva. A causa é desconhecida.
Síndrome de Bastian . Síndrome da seção completa da medula espinhal. Inclui uma fase inicial aguda bilateral, com paraplegia e anestesia abaixo do nível da lesão, arreflexia cutânea e tendinosa, retenção urinária e incontinência fecal; e uma fase posterior caracterizada por rigidez espástica e recuperação dos reflexos e tônus muscular. A causa é quase sempre traumática.
Síndrome de Bazex . Síndrome de acroceratose paraneoplásica. Consiste em lesões hiperceratóticas, com fundo eritematoso, em pavilhão auricular, nariz e dedos, acompanhadas de espessamento das unhas e perionixia. Geralmente, há uma neoplasia das vias aéreas superiores.
Síndrome de Behçet . Síndrome vasculítica com manifestações oculares e mucosas. Quadro recorrente, de evolução crônica, caracterizado por episódios de conjuntivite, uveíte e ulcerações de córnea e mucosa, bucal e genital. A origem é provavelmente viral e nas formas graves leva à cegueira.
Síndrome de Benedikt . Síndrome neurológica do tronco encefálico com envolvimento do terceiro nervo craniano e manifestações motoras e cerebelares. É um quadro de paralisia da motilidade ocular do lado da lesão (III nervo craniano), com hemiplegia frustrada, hernitembloro, hemiataxia e hemiasinergia do lado oposto (lesão do núcleo, vermelho e da via corticoespinhal). É produzida por hemorragias, amolecimento e tumores que acometem a região peduncular.
Síndrome de Bernard-Soulier . Síndrome hereditária relacionada a anormalidades no mecanismo de coagulação. É caracterizada por uma tendência à hemorragia acompanhada de trombocitopenia leve, plaquetas gigantes ou morfologia atípica, tempo de coagulação prolongado e consumo anormal de protrombina.
Síndrome de Bemhardt-Roth . Síndrome de Meralgia parestésica. É manifestada por parestesias e outros distúrbios sensoriais geralmente leves na face lateral de uma das coxas. É devido à constrição do nervo femoroccutâneo no nível de sua entrada na fáscia lata.
Síndrome de Bertolotti . Síndrome relacionada a anormalidades estruturais da coluna vertebral. É uma tríade composta por escoliose, sacralização da quinta vértebra lombar e ciática, as duas primeiras entendidas como anomalias: a básica e a terceira como complicação secundária.
Síndrome de Bianchi . Síndrome neurológica devido a lesão do córtex parietal. Afasia sensorial, apraxia e alexia, caracterizada pela incapacidade do paciente de compreender o significado das palavras que ouve ou lê, como se correspondessem a uma língua desconhecida. É típica de certas lesões, quase sempre tumorais, do lobo parietal esquerdo.
Síndrome de Bloch-Sulzberger . Síndrome do distúrbio do metabolismo do pigmento com múltiplas anormalidades corporais. É caracterizada por lesões cutâneas pigmentadas, vesiculares e posteriormente verrucosas, associadas a defeitos no desenvolvimento dos olhos, ossos e sistema nervoso central. É hereditário e afeta quase exclusivamente mulheres.
Síndrome de Blount-Barber . Síndrome relacionada a anormalidades estruturais dos membros . mais baixo. Associação de osteocondrose da epífise superior da tíbia, rotação interna da tíbia no fêmur e pés planos moderados. É devido ao desenvolvimento anômalo, de natureza isolada, da extremidade proximal da tíbia.
Síndrome de Bogorad . Síndrome que surge no período de recuperação de certos casos de paralisia facial. O distintivo é o aparecimento de lacrimejamento unilateral, do lado da paralisia, que ocorre quando o indivíduo ingere alimentos. É atribuída a peculiaridades do processo de regeneração nervosa, durante o qual algumas das fibrilas destinadas às glândulas salivares se conectariam com outras direcionadas ao sistema lacrimal.
Síndrome de Bonnet . Síndrome neurológica da base do crânio. Manifesta-se por oftalmoplegia completa, miose e distúrbios motores e sensoriais do nervo trigêmeo, de início insidioso e avanço progressivo. É produzida por infiltração de tumores na base do crânio.
Síndrome de Bonnier . Síndrome neurológica decorrente de lesão de estruturas centrais com envolvimento dos nervos cranianos III, V, VIII, IX e X. Manifesta-se por estado vertiginoso, fraqueza e sonolência, nevralgia no território trigeminal, alterações da motilidade ocular, instabilidade e hipoacusia e outros alterações. É produzida por lesões que afetam o núcleo, Deiters, as vias vestibulares associadas e os nervos cranianos III, V, VIII, IX e X.
Síndrome de bradiarritmia-taquiarritmia . Síndrome de períodos alternados de bradiarritmia e taquiarritmia. É caracterizada por períodos alternados de bradiarritmia (30 a 50 batimentos / min.) Com tontura ou síncope, que pode durar de minutos a meses, e períodos de taquiarritmia supraventricular (até. 200 por minuto), que pode durar de minutos a meses e ser acompanhada por palpitações e outros sintomas de taquicardia supraventricular paroxística. Nos intervalos entre eles, pode haver um ritmo sinusal normal. A causa é desconhecida.
Síndrome de Brissaud-Marie . Síndrome de histeria neurológica. Hemiespasmo de lábio e língua, sem causa justificativa, sem sistematização neurológica e em personalidade com traços histéricos.
Síndrome de Brissaud-Sicard . Síndrome 'causada por lesões irritativas do nódulo. Espasmo dos músculos faciais do lado da lesão central, com hemiplegia ou hemiparesia do lado oposto à lesão.
Síndrome de Bristowe . Síndrome do tumor do corpo caloso. É caracterizada por hemiplegia progressiva no lado da lesão, hemiparesia contralateral leve, disfagia, estupor, letargia, coma e morte.
Síndrome de Brown-Séquard . Síndrome de hemissecção da medula espinhal. Paralisia flácida, atrofia muscular e abolição da sensação profunda do lado da lesão, com preservação da sensação superficial, associada à perda da sensação superficial com preservação das habilidades motoras e sensação profunda do lado oposto ao da secção. A causa é quase sempre traumática (projéteis de armas de fogo).
Síndrome de Bruns . Síndrome neurológica de tumores do quarto ventrículo. É caracterizada por cefaleia intensa e intermitente, vertigem, vômitos e distúrbios visuais, que se acentuam quando o paciente faz um movimento brusco da cabeça.
Síndrome de Budd-Chiari . Síndrome de obstrução das veias supra-hepáticas. A forma aguda é caracterizada por dor epigástrica, hepatomegalia dolorosa rapidamente progressiva, ascite grave e icterícia leve, e leva à morte em alguns dias ou 1 a 2 meses. Na forma crônica, o início é insidioso e as manifestações acentuam gradativamente a hepatomegalia com grande esplenomegalia, dor abdominal, ascite e edema de membros inferiores. A obstrução é causada por trombose (endoflebite, policitemia) ou compressão extrínseca (por exemplo, hipernefroma).
Síndrome bulbar . veja Síndrome de Dejerine , def 2.
Síndrome bulbar hernilateral . veja Síndrome de Babinski-Nageotie .
Síndrome bulbar lateral . veja síndrome de Wallenberg .
Síndrome de Burnett . veja síndrome do leite-álcali .
Síndrome de Bumier . Síndrome endócrina e neurológica de origem tumoral. É caracterizada por nanismo, distrofia adiposogenital e atrofia do nervo óptico e geralmente é causada por tumores de crescimento lento que comprimem a hipófise e a região do quiasma.
Síndrome de Bywaters . veja síndrome de esmagamento .
Síndrome de Camurati-Engelmann . Síndrome da displasia diafisária progressiva . É caracterizada por espessamento simétrico e aumento do diâmetro da diáfise, dos ossos longos, com dor nas áreas afetadas, fadiga, marcha anormal e atrofia muscular. A causa é desconhecida.
Síndrome de Canadá-Cronkite . Síndrome da polipose intestinal com alterações orgânicas secundárias. É caracterizada pela presença de uma polipose intestinal. multiplex, levando à enteropatia crônica com esteatorreia. Como consequência, devido a múltiplas deficiências de nutrientes, ocorrem alopecia, alterações cutâneas e atrofia ungueal. A causa é desconhecida.
Síndrome do canal condilar . Síndrome neurológica da base do crânio. Paralisia unilateral da língua produzida pela compressão do nervo XII na base do crânio, geralmente de origem tumoral
Síndrome de Caplan . Síndrome artrítica associada a pneumoconiose. Ele define a associação de artrite reumatoide com a presença de múltiplos nódulos pulmonares (granulomas reumatoides) em pacientes com pneumoconiose subjacente. O distúrbio dificulta o transporte de gases através da parede alveolar.
Síndrome capsulotalâmica . Síndrome neurológica decorrente de lesão de estruturas subcorticais. É caracterizada por hemianopia, hemianestesia, hemiplegia parcial, alterações na percepção da dor e instabilidade emocional. É causada por lesões vasculares ou tumores do tálamo óptico e da cápsula interna.
Síndrome carcinóide maligna . Síndrome do tumor carcinóide do intestino delgado. É tipicamente caracterizada por episódios paroxísticos de rubor cianótico da face e pescoço, cólicas abdominais, diarreia, hipotensão e espasmo brônquico; e em estágios mais avançados devido a alterações pigmentares, ascite, lesões endocárdicas e insuficiência cardíaca. Geralmente, há várias metástases hepáticas e a condição se deve à hipersecreção de serotonina pelas células tumorais.
Síndrome centromedular . Síndrome neurológica devido a lesão da medula cervical. É caracterizada por paresia variável de todos os quatro membros, mas desproporcionalmente maior nos membros superiores. É causada por lesões da porção central da medula cervical, de origem vascular ou degenerativa.
Síndrome da medula espinhal centro-posterior . Síndrome neurológica devido a lesão medular. Manifesta-se por distúrbios vasomotores e dissociação da sensibilidade do tipo siringomélico (perda da sensibilidade térmica e dolorosa, com preservação da sensibilidade tátil e profunda). É produzida por lesões, da porção posterior central da substância cinzenta da medula espinhal
Síndrome pós-traumática do cérebro . Síndrome residual longe de traumatismo craniano. A condição é predominantemente polimórfica subjetiva, mas é frequente neste tipo de paciente e se manifesta por cefaleia de intensidade e localização variáveis, amnésia, vertigem, zumbido; palpitações, fadiga, irritabilidade, insônia e dificuldade de concentração.
Síndrome cervical . Síndrome da dor relacionada a alterações orgânicas da coluna cervical. É caracterizada por apresentar periodicamente dores cervicais, com irradiação para as costas, ombro e / ou membro superior. Pode ser simétrica ou unilateral e geralmente é exacerbada por movimentos de rotação ou extensão da cabeça. É devido à compressão ou irritação das raízes nervosas cervicais por impacto artrítico.
Síndrome de Cestan . Síndrome neurológica devido a extensos danos às estruturas cerebrais. Hemiplegia e hemianestesia no lado oposto da lesão, com hemiasinergia, laringoplegia, lateropulsão e síndrome de Horner no mesmo lado da lesão. A imagem se deve a lesões dispersas que afetam a pirâmide, as vias sensoriais, o pedúnculo cerebelar inferior, o núcleo ambíguo e o centro oculopupilar.
Síndrome da cimitarra . Síndrome radiológica causada pela boca anômala das veias pulmonares. É observada nas radiografias frontais de tórax e consiste em uma modificação da imagem cardíaca, que se torna convexa à direita (devido à abertura das veias pulmonares direitas na veia cava inferior) e côncava à esquerda (devido a deslocamento do coração para o pulmão direito, que é hipotrófico). No geral, a imagem lembra a de uma cimitarra, em que a lâmina é representada pelo coração e o cabo pelos grandes vasos.
Síndrome de Claude Bernard-Horner . Síndrome de paralisia cervical simpática. É caracterizada por subsidência do globo (enoftalmia), ptose da pálpebra superior com discreta elevação da pálpebra inferior e estreitamento da fissura palpebral, miose, anidrose e congestão vascular do lado afetado da face. A causa geralmente é tumor (compressão).
Síndrome de Claude-Lhermitte . Síndrome neurológica devido à compressão do infundíbulo e do terceiro ventrículo. É manifestada por um quadro polimórfico complexo, que inclui narcolepsia ou hipersonia, alterações vasomotoras e da regulação térmica, diabetes insípido e uma variedade de sintomas adicionais. A causa geralmente é tumor.
Síndrome do sopro sistólico clique final . Síndrome do prolapso da válvula mitral. É caracterizada pela ausculta de um clique sistólico tardio, na meso ou final da sístole, seguido de sopro sistólico agudo, crescendo-decrescendo, devido à regurgitação por um dos folhetos mitrais. O paciente pode apresentar sintomas (episódios de taquicardia, palpitações, dor torácica indefinida, etc.) ou não. É um distúrbio geralmente benigno que predomina em mulheres jovens.
Síndrome de Clouston . Síndrome de displasia ectodérmica hidrótica. É caracterizada por hiperceratose palmoplantar, hiperpigmentação da pele, hipotricose, destruição das unhas e, frequentemente, retardo mental. A transmissão é autossômica dominante.
Síndrome de coagulação intravascular disseminada . Síndrome associada a trombose e hemorragia disseminada ao nível da microcirculação. É uma imagem de microtrombose generalizada acompanhada por hemorragias napa, frequentemente irreversível, o que é devido à intervenção de vários mecanismos de sobreposição: o consumo e esgotamento de factores de coagulação, danos generalizados para o endotélio capilar, fibrinólise exagerada, choque, etc. O processo é acompanhada por trombocitopenia, fibrinogenpenia e excesso de produtos de degradação da fibrina na corrente sanguínea. É observada em queimaduras graves e pessoas feridas, no choque séptico, na leucemia e em outras condições de gravidade semelhante.
Síndrome de Cogan ., Síndrome associada a distúrbios de visão rápida e audição. Presença de ceratite intersticial reversível não sifilítica, acompanhada de vertigem, zumbido, nistagmo e surdez irreversível de evolução rápida. Afeta adultos jovens e a causa é desconhecida.
Síndrome de Cauda equina . Síndrome neurológica devido à compressão das raízes lombares e sacrais . Paraplegia flácida, arreflexia tendinosa, anestesia em sela dolorosa (anal, perineal, genital e glútea), distúrbios esfincterianos e impotência. É produzida por trauma ou compressão tumoral.
Síndrome da cólera pancreática . veja Síndrome de WDHA .
Síndrome IBS . Síndrome de uma condição crônica benigna do intestino grosso. É caracterizada por alterações da motilidade intestinal, com cólicas e períodos de constipação e diarreia, sendo geralmente exagerada a secreção de muco. É considerado um distúrbio funcional.
Síndrome. de Collet-Sicard . Síndrome neurológica da base do crânio. O quadro é idêntico ao da síndrome de Villaret (lesão unilateral dos nervos cranianos IX, X, XI e XII), mas sem paralisia simpática cervical. As: causas também são as mesmas.,
Síndrome de compressão da artéria vertebral . veja Síndrome de Vertigem Cervical
Síndrome do canal auditivo interno . Síndrome neurossensorial de lesão do nervo auditivo. Surdez de percepção, hipoestesia e hiporreflexia corneana ou arreflexia vestibular. A causa mais frequente é o neurinoma do nervo auditivo.
Síndrome do ducto cístico remanescente . veja Síndrome do Coto Cístico
Síndrome do Ducto Lombar Estreito . Síndrome neurológica periférica devido à irritação das raízes nervosas. É caracterizada por dor lombar acompanhada de radiculalgia crural ou ciática, que aparece ou se torna muito intensa ao caminhar (claudicação radicular intermitente); ocasionalmente, há parestesias nos pés, paresia muscular e distúrbios esfincterianos discretos. É causada pelo estreitamento congênito do ducto lombar, mas geralmente se manifesta em geral após os 50 anos, devido ao acréscimo de outros processos patológicos (osteofitose, etc.).
Síndrome do cone medular . Síndrome neurológica das lesões do segmento terminal da medula. Anestesia pudendária, às vezes em uma distribuição em "sela", incontinência urinária e fecal devido à disfunção do esfíncter, abolição da ejaculação e ereção e perda dos reflexos anal e plantar, e a causa geralmente é tumor ou traumática.
Síndrome do Coração Pulmonar Aguda . Síndrome da insuficiência circulatória aguda de origem pulmonar. Ocorre abruptamente e se manifesta como dispneia intensa, cianose ou lividez, hipertensão venosa com ingurgitamento jugular e hipotensão arterial sistêmica, dilatação da artéria pulmonar e deslocamento do eixo elétrico do ECG para a direita. A mortalidade é alta e é causada por embolia ou trombose da artéria pulmonar ou de seus ramos.
Síndrome do Coração Pulmonar Crônico . Síndrome de insuficiência cardíaca direita devido à hipertensão prolongada da pequena circulação em neuropatias crônicas. É caracterizada por dispneia aos esforços, com ou sem crises de asma, tosse crônica, baqueteamento digital, cianose, cefaleia e sonolência; nos estágios avançados, hepatomegalia congestiva e edema periférico. Há poliglobulia, sinais radiológicos de doença pulmonar subjacente e alterações eletrocardiográficas típicas (onda P alta e pontiaguda em II, III e aVF e negativa em aVL, onda R alta em VI etc.). É visto no enfisema, bronquite crônica e bronquiectasia, tuberculose pulmonar e outras condições.
Síndrome do cordão anterior . Síndrome neurológica devido a lesão medular. A lesão localiza-se na porção anterior da medula espinhal, havendo hipoalgesia, hipoestesia e paralisia completa abaixo desse nível, com relativa preservação das funções que dependem da medula posterior (sensibilidade tátil, vibratória e posicional). A causa geralmente é traumática (projéteis de arma de fogo).
Síndrome coreica . Síndrome neurológica por lesão dos núcleos cinzentos da base. Movimentos musculares anormais, amplos, anárquicos, involuntários e irreprimíveis, como os de uma dança estranha, que não obedecem a nenhuma sistematização e que desaparecem durante as horas de sono; está associada à hipotonia muscular. Existe uma forma crônica hereditária progressiva (carea de Huntington) e uma forma aguda que acompanha a gravidez ou certos casos de febre reumática (carea de Sydenham). É causada por lesão dos núcleos cinzentos da base (caudado, putâmen, pallidum, centro de Luys e locus niger).
Síndrome coronariana intermediária . Nome geral das imagens de isquemia coronariana que não correspondem a um infarto do miocárdio, nem se enquadram no padrão característico da angina de peito. Inclui três sintomas diferentes: a) a síndrome da insuficiência coronariana aguda, que se caracteriza pelo aparecimento isolado de um episódio doloroso intenso, muitas vezes em repouso, acompanhado de alterações eletrocardiográficas: típicas: na onda T e no segmento ST, mas sem modificações no complexo QRS; b) a síndrome da angina instável semelhante à anterior, mas definida pela apresentação de episódios repetidos durante o repouso; ec) a síndrome da angina progressiva ou pré-infarto, em que os episódios aparecem em resposta a esforços cada vez menos intensos;ou com mais; frequência, ou com maior duração, ou com modificação qualitativa de suas características.
Síndrome de costela do enfisema pulmonar . Síndrome da dor associada à doença pulmonar obstrutiva crônica. Dor constante que o paciente refere ao perímetro da base do tórax, e que geralmente se intensifica durante as complicações usuais desta condição (bronquite aguda; etc.). É atribuída a fatores estruturais (hiperexpansão da caixa torácica, osteoporose senil, etc.) e ao esforço muscular permanente que o indivíduo faz para respirar.
Síndrome de Costen . Síndrome da disfunção temporomasticatória. É caracterizada por dor na região temporomandibular, acompanhada de ranger da articulação correspondente, lateralização da mandíbula ao abrir a boca e, ocasionalmente, travamento da mandíbula; Também pode haver sintomas auditivos (dor, perda de audição, zumbido). É devido à má oclusão da articulação temporomandibular, com irritação da corda do tímpano e dos nervos auriculotemporais.
Síndrome da costela cervical . veja Síndrome do tórax estreito superior .
Síndrome costoclavicular . veja Síndrome do tórax estreito superior .
Síndrome de Courvoisier-Terrier . Síndrome do carcinoma de Vater ampulla. O quadro é caracterizado por icterícia, acólia e colúria, associadas a acentuada dilatação da vesícula biliar, mas sem a rápida caquexia que acompanha o carcinoma da cabeça do pâncreas.
Síndrome CREST. Síndrome associada a esclerodermia. O seu nome deriva das principais manifestações do quadro: C de calcose, devido à presença de depósitos de cálcio dispersos ao nível subcutâneo e periarticular; R de Raynaud, para a frequência com que o fenômeno de Raynaud aparece nos estágios iniciais da doença; ES para esclerodactilia, devido ao predomínio nos dedos da rigidez que atinge todo o corpo, e T para telangiectasia, devido ao desenvolvimento deste tipo de anormalidades na superfície da pele. Geralmente é observada nas formas menos graves de esclerose sistêmica progressiva.
 Síndrome de Crigler-Najjar Síndrome de Crigler-Najjar
|
Síndrome de Crigler-Najjar . Síndrome neurológica relacionada a anormalidades congênitas no metabolismo da bilirrubina. É caracterizada pelas manifestações da impregnação ictérica dos núcleos da base (kernicterus) (distúrbios neurológicos graves), acompanhada por níveis elevados de bilirrubina não conjugada no sangue. A doença é evidente desde o nascimento, geralmente leva à morte durante o primeiro ano de vida e a causa é uma deficiência hereditária de glucuroniltransferase,
Síndrome de Cruveilhier-Baumgarten . Síndrome de hipoplasia congênita do sistema venoso supra-hepático. O quadro é causado pela oclusão dos ramos intra-hepáticos das veias indicadas, e suas manifestações, que são graves, decorrem de dano orgânico direto causado pela malformação (doença fibrosa crônica do fígado); a repercussão hemodinâmica resultante (hipertensão portal, com varizes esofágicas, notáveis esplenomegalia e hiperesplenismo); da função compensatória exercida pelas veias umbilicais, que não colapsam ao nascimento (circulação colateral em direção à veia cava superior, sopro suave, contínuo, auscultável na região periumbilical) e de eventuais complicações (insuficiência hepática, sangramento digestivo, etc. )Ao contrário de outros processos com características semelhantes, não é acompanhado de ascite.
Síndrome do corpo caloso , veja síndrome de Bristowe .
Síndrome da pele paraneoplásica . Síndrome de pele associada a processos tumorais em outras regiões do corpo. Inclui uma variedade de condições que precedem ou acompanham o aparecimento das manifestações clínicas de um tumor, incluindo acantose nigricans (câncer gástrico e, menos frequentemente, tumores intestinais, pulmonares ou ginecológicos), dermatomiosite (neoplasias mamárias ou genitais e, às vezes, digestivas ou respiratório), ictiose adquirida (doença de Hodgkin e condições hematopoéticas malignas), acroqueratose de Bazex (câncer da laringe) e assim por diante.
 Síndrome de Chotzen |
Síndrome de Cyriax . Síndrome dolorosa das cartilagens costais. É caracterizada por dor de intensidade variável na região paraesternal que se irradia com relativa frequência para o pescoço, ombro e / ou braço. O quadro é semelhante ao da angina de peito, do qual difere por ser persistente e por se agravar quando se palpam uma ou mais cartilagens costais, onde está a causa do problema.
Síndrome de Charlin . Síndrome de neuralgia do nervo nasal. Suas características são: 1) dor intensa, unilateral, nas crises paroxísticas, em região nasoorbital, 2) hidrorreia ipsilateral e 3) lesões tróficas da córnea. É devido à neurite causada por doenças do nariz.
Chauffard-Still síndrome ver de Ainda síndrome .
Síndrome de Chiari-Frommel . Variedade de síndrome de galactorreia não puerperal. É típica do pós-parto muito distante e é caracterizada por lactação prolongada, amenorréia e atrofia genital, no primeiro caso devido à hipersecreção sustentada de prolactina, e nos dois casos restantes devido à diminuição concomitante na secreção de gonadotrofinas hipofisárias e como um conseqüência disso, de estrogênios pelo ovário. A causa não é conhecida com precisão, mas está localizada na própria hipófise ou no hipotálamo.
Síndrome de Chotzen . Síndrome hereditária caracterizada por malformações predominantemente cranianas e digitais . Designa a combinação de acrocefalia com polidactilia e sindactilia parcial, à qual às vezes se somam malformações faciais (hipertelorismo ocular) e retardo mental. A condição é transmitida como um traço autossômico dominante.
Síndrome de Christ-Siemens . Síndrome da displasia ectodérmica anidrótica. É caracterizada por: a) pele lisa e lustrosa, ausência de glândulas sudoríparas e má formação de pelos; b) nariz em sela e testa e queixo proeminentes; c) ausência de gosto e cheiro; d) anomalias dentais ee) retardo mental. É transmitido como um traço ligado ao X.
 Síndrome de Churg-Strauss |
Síndrome de Churg-Strauss . Síndrome da granulomatose alérgica. Eosinofilia proeminente, infiltrados pulmonares difusos e associação com asma brônquica. É uma vasculite semelhante. ao da poliarterite nodosa, que afeta também o coração, os rins, o intestino e os nervos periféricos.
Síndrome de Da Costa . Síndrome de astenia neurocirculatória. Complexo de sintomas crônicos 'caracterizado por asfixia, tontura, sensação de cansaço, dor no peito e palpitações, sem alterações orgânicas cardíacas. É de natureza emocional e também é chamado de coração de soldado.
Síndrome de Dandy-Walker . Síndrome neurológica resultante de um obstáculo congênito. na circulação do líquido cefalorraquidiano. É manifestada desde o nascimento por hipertensão intracraniana, hidrocefalia e distúrbios predominantemente cerebelares. A causa é a atresia congênita do forame de Magendie e Luschka do quarto ventrículo, que impede a drenagem normal do líquido cefalorraquidiano.
Síndrome de Debré-Sémélaigne . Síndrome de cretinismo hipotireoidiano com distúrbios musculares. A associação de cretinismo hereditário aumenta o volume muscular e diminui a contração dos músculos e reflexos tendinosos. É de causa genética e é transmitida como um traço autossômico recessivo.
Síndrome de deficiência de ácido nicotínico . Síndrome da deficiência de um nutriente essencial. Na sua forma típica (pelagra) é caracterizada por manifestações digestivas (diarreia, náuseas e vômitos), pele: (pigmentação facial, eritema da base do pescoço) e membranas mucosas (atrofia das papilas; lingual, escarlate, áspera e língua fissurada), e em casos graves, devido a distúrbios neurológicos. O distúrbio é raro, mas pode ser observado em pacientes com câncer ou diarreia crônica.
Síndrome de deficiência de adenosina desaminase . Síndrome de imunodeficiência congênita com anormalidades osteocartilaginosas graves. Ela se manifesta nos primeiros 6 meses de vida por: a) vômitos, diarréia e parada do desenvolvimento; b) anomalias metafisárias, deformação em taça das junções costocondrais e membros curtos; ec) infecções recorrentes graves; predominantemente respiratório, cutâneo e digestivo, causado por vírus, bactérias, fungos ou protozoários. A linfopenia é observada no sangue circulante, com uma diminuição acentuada nos linfócitos T e, às vezes, também nos linfócitos B.
Síndrome de deficiência de imunoglobulina M isolada . Síndrome de imunodeficiência hereditária. Manifesta-se por baixos níveis de IgM circulante, com distúrbios atópicos e gastrointestinais, esplenomegalia e tendência ao desenvolvimento de infecções recorrentes graves; e processos malignos. O transtorno é familiar e quatro vezes mais comum em homens do que em mulheres.
Síndrome de deficiência de aldolase hepática . Síndrome produzida por uma anormalidade congênita no metabolismo da frutose. É caracterizada por parada precoce do desenvolvimento, tendência acentuada à hipoglicemia, eliminação de frutose na urina, distúrbios renais e hepáticos que, em casos graves, terminam em cirrose com ascite e esplenomegalia. A causa é uma deficiência congênita de aldolase no fígado.
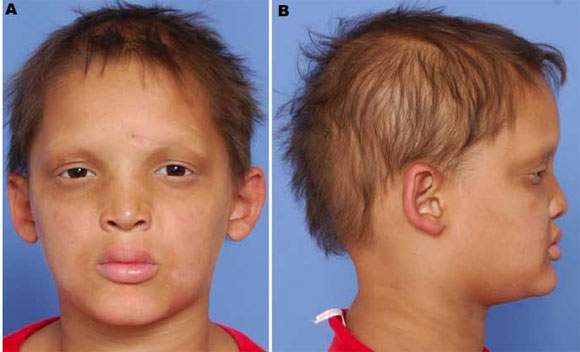 Síndrome de Christ-Siemens |
Síndrome de deficiência de fosforilase-b-quinase . Síndrome hereditária com distúrbios hepáticos e manifestações hipoglicêmicas. É caracterizada por hepatomegalia, hipoglicemia de jejum e retardo de crescimento. É uma doença benigna transmitida como traço ligado ao X e cujas manifestações geralmente desaparecem na adolescência.
Síndrome de deficiência de ferro . Síndrome da deficiência de um nutriente essencial. Inclui manifestações gerais (astenia, fadiga, hipersensibilidade ao frio, etc.), alterações mucocutâneas; (atrofia da papila lingual, estomatite angular, pele seca, etc.), e principalmente palidez e anemia com microcitose e hipocromia, diminuição do ferro sérico, aumento da capacidade de combinar ferro e ausência desse mineral na medula óssea estendida. É causada por deficiências absolutas ou relativas (dieta láctea, gravidez múltipla), por perda exagerada (hemorragia gastrointestinal oculta, hipermenorreia) ou por uma combinação destes mecanismos.
Síndrome de deficiência purina nucleosídeo fosforilase . Síndrome de imunodeficiência congênita com anormalidades hematológicas e do tecido conjuntivo. A condição não aparece ao nascimento, mas após o primeiro ano de vida, principalmente devido a infecções recorrentes associadas a uma diminuição progressiva dos linfócitos T circulantes e, às vezes, com alterações nos tecidos de suporte e anemia hemolítica. É hereditário e transmitido como traço autossômico recessivo.
Síndrome de deficiência de vitamina A . Síndrome da deficiência de um nutriente essencial. É caracterizada por alterações cutâneas (queratose, xeroderma, hiperqueratose folicular), distúrbios oculares (xeroftalmia, astenopia, dificuldade de visão noturna) e facilidade de contrair infecções, principalmente do trato respiratório. A condição raramente é vista em sua forma pura, devido à abundância de vitamina A nos alimentos, mas é vista como um componente de estados graves de desnutrição.
Síndrome de deficiência de vitamina B . (tiamina). Síndrome de deficiência de nutrientes específicos. Dá origem ao beribéri e inclui manifestações gerais (astenia, anorexia, etc.), distúrbios neuromusculares (fraqueza dos membros inferiores; polineurite; com arreflexia ou hiporreflexia patelar e parestesias, etc.) e alterações cardiovasculares na variante úmida da condição (cardiomegalia, insuficiência cardíaca congestiva, etc.). É vista como um componente de sintomas multicarenciais, com alguma frequência na desnutrição em alcoólatras e, ocasionalmente, em bebês e na gravidez.
Síndrome de deficiência de vitamina B 2 (riboflavina) . Síndrome da deficiência de um nutriente essencial. Manifesta-se por alterações cutâneas (queilite, estomatite anular; disfunção sebácea, etc.) e das membranas mucosas (glossite, atrofia da papila lingual) e por distúrbios oculares (astenopia, ambliopia, vascularização da córnea). É observado, quase sempre, 'como um componente de tabelas multidiferenciais.
Síndrome de deficiência de vitamina B 6 (piridoxina) . Síndrome da deficiência de um nutriente essencial. Inclui alterações mucocutâneas. (glossite seborréia nasolabial), distúrbios neurológicos (neuropatia periférica, que predomina nos membros inferiores) e, em certos casos da infância, convulsões resistentes aos medicamentos usuais. Quase sempre é visto como um componente de quadros de multi-trader.
Síndrome de deficiência de vitamina B 12 (cianocobalamina) . Síndrome da deficiência de um nutriente essencial. Dá origem ao quadro de anemia perniciosa, que inclui alterações cutâneas e mucosas (palidez, icterícia leve amarelo-limão, glossite), manifestações neurológicas (parestesias, ataxia, arreflexia tendínea, nemite óptica) e anormalidades hematológicas (anemia hipercrômica, megalocitose, presença de megaloblastos na medula óssea). É causada por um suprimento insuficiente da vitamina ou pela falta de fator intrínseco gástrico (essencial para sua absorção), seja como consequência de uma gastrectomia ou de origem constitucional (com acólia e hipocloridria).
Síndrome de deficiência de vitamina C (ácido ascórbico) . Síndrome da deficiência de um nutriente essencial. A forma clássica é o escorbuto e se manifesta por fadiga e distúrbios emocionais, inchaço e sangramento gengival e dores musculares em adultos; e em crianças devido à dor e inchaço dos membros inferiores, alterações radiológicas dos ossos (hemorragias subperiosteais, aspecto de vidro fosco, etc.), inchaço e sangramento gengival e sangramento em outras regiões do corpo. Atualmente é uma deficiência muito rara, que pode afetar indivíduos que vivem sozinhos e, ocasionalmente, bebês.
Síndrome de deficiência de vitamina D (colecalciferol). Síndrome da deficiência de um nutriente essencial. Manifesta-se, na criança, pelo complexo sintomático de raquitismo (craniotabes, dentição retardada, deformações pélvicas e da caixa torácica, curvatura anormal das pernas, etc.); e em adultos para tetania e osteomalácia que predominam no crânio, pelve; e joelhos. O quadro é observado em casos de deficiência alimentar (com produção endógena insuficiente) de perda exagerada (síndromes de má absorção) ou em raquitismo resistente à vitamina D (insuficiência renal crônica, distúrbios tubulares renais, etc.).
Síndrome de deficiência de vitamina K (menadiona). Síndrome da deficiência de um nutriente essencial. No recém-nascido, manifesta-se por hematomas cranianos ou esternocleidomastóideos, hemorragias meníngeas, orais ou do cordão umbilical e lesões purpúricas; Aparece dois ou três dias após o nascimento e é devido à incapacidade de absorver vitamina K devido à ausência de flora intestinal normal. Em adultos, é evidenciada por petéquias, equimoses, hematomas ou hemorragia, e geralmente é consequência do uso terapêutico de anticoagulantes cumarínicos.
Síndrome de Dejean . Síndrome neurophtálmica devido a condições do assoalho orbital. Inclui exoftalmia e diplopia devido ao deslocamento do globo ocular; dor intensa na região da mandíbula superior e dormência na área inervada pelos dois primeiros ramos do trigêmeo (oftálmico e maxilar superior). A causa geralmente é tumor.
Síndrome de Déjerine . Síndrome neurológica das lesões do córtex sensorial. Manifesta-se pela perda de uma variedade de funções altamente especializadas, de forma que o paciente é incapaz de reconhecer um objeto conhecido pelo toque (astereognosia), de reconhecer mudanças na posição de segmentos corporais ou diferenças na intensidade dos estímulos, etc. É causada por lesões vasculares (embolia) e às vezes por tumores. // 2. Síndrome neurológica associada a lesões focais da porção anterior do bulbo.Do lado oposto ao da lesão, ocorre hemiplegia que respeita a face, enquanto do lado da lesão observa-se paralisia e atrofia da língua (nervo XIT) quando a lesão é superior, ou paralisia do palato mole e vogal do cordão (nervo X) quando localizada em um nível.
Síndrome de Déjerine-Klumpke . Síndrome da paralisia braquial de tipo inferior. É uma paralisia atrófica dos músculos da mão e dos dedos, associada à síndrome de Claude. Bernard-Homer. É devido à lesão das raízes C8 e DI, geralmente devido à compressão.
Síndrome de Déjerine-Sottas . Síndrome de polineuropatia hipertrófica progressiva. Manifesta-se inicialmente por dor e parestesia nos pés e, posteriormente, por fraqueza e atrofia dos segmentos distais dos membros, arreflexia tendínea e incapacidade precoce. Os nervos estão um tanto espessados pelo depósito exagerado de material de colágeno. O distúrbio é herdado e transmitido como um traço autossômico recessivo.
Síndrome de Del Castillo-Trabucco-de la Balze . Síndrome associada à ausência congênita de epitélio germinativo. É caracterizada por esterilidade e testículos pequenos, com libido normal e características sexuais secundárias. O testículo contém células normais de Leydig e Sertoli, mas nenhuma célula germinativa.
Síndrome de Dercum . Síndrome de adipose dolorosa . Presença de depósitos de gordura circunscritos e dolorosos no tecido subcutâneo das extremidades e, às vezes, em outros setores do corpo. Geralmente é de aparência esporádica, com alguns casos documentados de incidência familiar.
Síndrome de descondicionamento cardiovascular . Síndrome ergométrica de indivíduos normais não treinados. Manifesta-se durante testes de esforço controlados por frequência cardíaca e pressão arterial que aumentam normal e muito rapidamente, sem alterações eletrocardiográficas significativas e com capacidade de esforço medíocre. Representa a condição ideal para indicar exercícios físicos
Síndrome de descerebrados . Síndrome neurológica devido à autonomia do tronco cerebral. É caracterizada por hiperextensão dos membros inferiores com espasticidade acentuada, flexão fixa dos braços na parede torácica, hiperextensão da cabeça, divergência ocular e coma em que apenas as funções vegetativas são mantidas. É devido a lesões que interrompem a condução nervosa ao nível dos tubérculos quadrigêmeos, de origem traumática ou tumoral ou devido a hemorragias do terceiro ventrículo.
Da síndrome de Toni-Debré-Fanconi . veja síndrome de Fanconi .
Síndrome de diarreia aquosa . veja Síndrome de WDHA .
Síndrome de Di Ferrante . Síndrome da mucopolissacaridose VIII Forma semelhante à das síndromes de Morquio ou Sanfilippo, com estatura e retardo mental, hepatomegalia e esplenomegalia discretas, perda auditiva e grânulos metacromáticos em leucócitos.
Síndrome de Dighton-Adair . Síndrome de Osteogenesis imperfecta. Suas características básicas são: a) osteoporose generalizada com acentuada fragilidade dos ossos, e com o tempo o desenvolvimento de múltiplas deformações ósseas produzidas pela osteomalácia e pela consolidação das fraturas; b) esclera azul ec) surdez por osteosclerose, em cerca de um terço dos casos. Os níveis de cálcio e fósforo no sangue e na urina são normais. A imagem é familiar e é transmitida como um traço autossômico dominante.
Síndrome de dilatação estomacal aguda . Síndrome pós-operatória complicada. Vômitos abundantes e contínuos, distensão abdominal rápida e salpicos progressivos e presença de grande nível de líquido na radiografia simples de abdome, realizada em pé. Ocorre em indivíduos submetidos à cirurgia intra-abdominal, quando não foram submetidos à aspiração gástrica por sonda.
Síndrome da mão desajeitada com disartria . Síndrome neurológica lacunar devido a lesão no joelho e no braço anterior da cápsula interna. É caracterizada por disartria, paralisia facial e paresia da língua, acompanhada por leve fraqueza e falta de jeito nos movimentos das mãos. A causa é vascular (trombose, embolia) no território lenticulostriado.
Síndrome do disco intervertebral . Síndrome neuromuscular devido à compressão da raiz. É caracterizada por dor geralmente intensa na região lombar, que surge durante ou logo após um intenso esforço de flexão ou hiperextensão do tronco nos membros inferiores, e que é caracteristicamente exacerbada por movimentos e manobras: que aumentam a pressão do cérebro fluido (tosse, defecação, etc.). A dor frequentemente irradia para o território do ciático (face posterior da coxa e porções laterais da perna e pé) e, em casos graves, é acompanhada por hiporreflexia ou arreflexia aquiliana e sinais de degeneração nervosa no eletromiograma. É devido à compressão de uma raiz nervosa pela protrusão de um disco intervertebral lombar.
Síndrome de disfagia e disfonia . Síndrome neurológica de lesão neumogástrica. É caracterizada por alterações unilaterais, incluindo paralisia do palato mole, perda do reflexo de vômito e paralisia das cordas vocais. É devido a uma lesão intracraniana, geralmente de origem tumoral.
Síndrome de disfunção placentária . Síndrome do sofrimento fetal intrauterino. É caracterizada por intensa pigmentação amarela da pinta sebácea, pele e unhas do feto, pigmentação esverdeada do cordão umbilical, associada ao gigantismo e muitas vezes à morte do feto antes do nascimento. É típico de gestações que duram mais de trezentos dias e é produzido pela degeneração das estruturas placentárias.
Síndrome da disfunção temporomasticatória . veja síndrome de Costen .
Síndrome da disgenesia gonadal . veja a síndrome de Turner
Síndrome de disceratose congênita . Síndrome hereditária caracterizada por alterações cutâneas e tendência ao desenvolvimento de processos malignos. É definida pela presença de hiperpigmentação reticular, hiperqueratose palmar e plantar, leucoplasia mucosa e perda de unhas; há também pancitopenia e alta tendência ao desenvolvimento de carcinomas. É transmitido como um personagem ligado ao sexo.
Síndrome de distrofia muscular pós-menopausa . Síndrome miopática de etiologia desconhecida É caracterizada por fraqueza progressiva da musculatura proximal dos membros inferiores e superiores, com dificuldade crescente em realizar movimentos e atividades normais.
Síndrome da distrofia simpática reflexa . Síndrome dolorosa acompanhada de distúrbios musculares, tróficos e vasomotores. Aparece em uma extremidade logo após uma situação patológica desencadeadora (infarto do miocárdio, trauma, condição neurológica) e se manifesta como edema distal (por exemplo, de uma mão inteira ou de um pé inteiro), dor intensa e em queimação na mesma região e labilidade vasomotora com episódios de vasoconstrição ou vasodilatação. Pode permanecer inalterado por dias ou semanas e depois reverter completamente ou evoluir progressivamente até produzir alterações tróficas graves, atrofia muscular e contratura em flexão. A causa não foi estabelecida com precisão, embora geralmente seja atribuída uma origem simpática reflexa.
Síndrome de Down . Síndrome genética caracterizada por retardo mental e anormalidades típicas. Suas características básicas são: a) baixa estatura e crânio pequeno, principalmente no diâmetro ântero-posterior; b) fácies característica com nariz pequeno, olhos oblíquos e língua grande e protuberante; c) dedo mínimo curto, separação excessiva entre os dois primeiros dedos das mãos e dos pés, e presença de dobras palmares simiescas; d) presença de anormalidades esqueléticas leves e, com bastante frequência, malformações cardiovasculares; ee) retardo mental leve a grave. É devido a uma trissomia do cromo soma 21, de aparência espontânea e mais raramente familiar.
Síndrome de Dresbach . Síndrome de eliptocitose hereditária . É caracterizada pela presença de eritrócitos ovais ou elípticos, com hemólise pouco acentuada, esplenomegalia leve e poucas manifestações clínicas. É transmitida de forma autossômica dominante e se deve a um defeito congênito no citoesqueleto eritrocitário.
Síndrome de Dressler . Síndrome da dor febril em certos pacientes com infarto do miocárdio. É manifestado por um quadro febril que aparece 1 a 6 semanas após um infarto do miocárdio e é acompanhado por dor retroesternal com características de dor pleuropericárdica: irradia para o pescoço e ambos os ombros, alivia quando o indivíduo inclina o tronco para frente, e é exacerbado pela inspiração profunda. A causa é a pleuropericardite, frequentemente associada à pneumonite, e é considerada uma reação auto-imune.
Síndrome de Duane . Síndrome congênita caracterizada por distúrbios oculares unilaterais. Manifesta-se pela impossibilidade ou dificuldade acentuada de rotação externa do olho, dificuldade de rotação interna, retração do globo ocular e convergência defeituosa. O distúrbio é hereditário e é transmitido como um traço autossômico dominante.
Síndrome de Duchenne . Síndrome neurológica de paralisia bulbar progressiva. É um quadro progressivo de disartria, disfonia e paralisia, precedido de espasmo dos músculos dos lábios e da língua, devido ao envolvimento crescente dos nervos cranianos V, VII; IX, X e Xll. A condição é de natureza degenerativa e sua causa é desconhecida.
Síndrome de dumping . veja Síndrome de esvaziamento rápido .
Síndrome de Duplay . Síndrome de tendinite calcária. Consiste em dor e limitação dos movimentos da articulação do ombro, devido à inflamação e calcificação da bursa subacromial ou subdeltoide.
Síndrome de Eaton-Lambert . Síndrome miastênica associada a certas neoplasias pulmonares. É manifestada por fraqueza das porções proximais dos membros superiores e inferiores, acompanhada por dor, dormência e arreflexia ou hiporreflexia tendínea. Ao contrário da síndrome da miastenia gravis, a) não há alterações da musculatura ocular eb) a força tende a aumentar, e não diminuir, com a repetição dos movimentos musculares. Em geral, a condição precede o aparecimento de um carcinoma de pulmão em meses ou até dois anos. células, pequenas.
Síndrome eclâmptica . Síndrome edematosa hipertensiva da gravidez. É manifestada pela tríade de hipertensão, edema e albuminúria, além de náuseas e vômitos e, eventualmente, manifestações de sobrecarga cardíaca e sinais neurológicos de hipertensão intracraniana, incluindo convulsões e coma. É típico dos últimos estágios da gravidez.
Síndrome de Eddowes . veja Síndrome Digihton-Adair
Síndrome de Ehlers-Danlos . Síndrome associada a doenças hereditárias do tecido conjuntivo. É caracterizada por hiperelasticidade e fragilidade da pele, frouxidão articular e facilidade para desenvolver hemorragias subcutâneas e hematomas musculares que posteriormente calcificam. O distúrbio é transmitido como um traço autossômico dominante.
Síndrome de Eisenmenger . Síndrome caracterizada por malformações cardíacas e hipertensão pulmonar. É definida pela presença de CIV alto, com impulso da aorta sobre o defeito septal, hipertrofia do ventrículo direito e dilatação da artéria pulmonar, evidenciada pelo alargamento do arco médio na radiografia de tórax frontal.
Síndrome da doença do nó sinusal Síndrome da disfunção do nó sinusal de etiologia desconhecida. Clinicamente, manifesta-se por crises temporárias de fraqueza, fadiga fácil, tontura e, às vezes, estados de síncope, que reaparecem com frequência variável em indivíduos sem sinais de doença cardíaca subjacente e que se devem a episódios súbitos de bradiarritmia (bradicardia sinusal, bloqueio sinoatrial , parada do seio nasal). O eletrocardiograma basal é geralmente normal, de modo que o diagnóstico só pode ser feito acidentalmente (ocorrência da anormalidade durante o registro) ou se o médico suspeitar e solicitar um registro Holter contínuo e prolongado.
Síndrome de Erb . veja Síndrome de Miastenia Gravis .
Síndrome do escaleno anterior . veja Síndrome do tórax estreito superior .
Síndrome do exercício . veja Síndrome de Da Costa .
Síndrome do espaço póstero-externo do côndilo . veja síndrome de Collet-Sicard .
Síndrome do espaço retroparotídeo . veja Síndrome de Villaret
Síndrome esquizofrênica . Síndrome psicótica caracterizada por um profundo distanciamento do indivíduo da realidade, baseado em graves alterações de personalidade, pensamento e afetividade. É observada na adolescência e na juventude e se manifesta, basicamente, por: a) distúrbios de personalidade, com perda do senso de individualidade e unicidade, e muitas vezes com a convicção de ser controlado por pessoas ou forças estranhas a ele; b) alterações de pensamento, pelas quais o indivíduo explica seu ambiente com base em fatos inconseqüentes dessa realidade, aos quais atribui um sentido arbitrário, e nas construções racionais rigidamente estruturadas que faz a partir desses fatos;c) obscurecer as formas de expressão, que as tornam enigmáticas e apenas compreensíveis para ele próprio; d) apatia, inércia, negativismo, estupor e, eventualmente, catatonia e autismo; e) insatisfação pronunciada ef) preservação da inteligência.
Síndrome da estenose superior do tórax . Síndrome neurovascular devido à irritação do plexo braquial. É um complexo sintomático referido ao membro superior ou a qualquer uma de suas partes, principalmente a mão, que se manifesta por dor, parestesia e fadiga; por alterações vasomotoras: locais (acrocianose, palidez, frieza, fenômeno de Raynaud e ocasionalmente edema leve) e eventualmente por alterações tróficas (ulcerações digitais) e atrofias musculares localizadas.É devido à irritação, compressão ou alongamento dos componentes do plexo braquial por estruturas vizinhas, e abrange condições de origem diferente, mas com consequências semelhantes: a) a síndrome do escaleno anterior, em que a artéria subclávia e o plexo braquial são comprimidos entre o escaleno anterior, a primeira costela e o escaleno mediano; b) síndrome da costela cervical,
Síndrome de exoftalmia maligna . Síndrome oftalmopática de tireotoxicose. Manifesta-se por uma exoftalmia bilateral pronunciada, com paresia dos músculos oculomotores externos, inchaço das pálpebras, edema e injeção da conjuntiva e dor retroocular constante. É típico da doença de Graves.
Síndrome de Fanconi . Nome geral do conjunto de anomalias que resultam de uma disfunção dos túbulos proximais. É observada como forma idiopática ou associada a uma variedade de processos patológicos (amiloidose, nefrose, intoxicação por tetraciclinas expiradas, etc.), e é definida com base nos seguintes critérios: a) pela presença de quatro anomalias básicas: glicosúria não associado a hiperglicemia, fosfatúria, aminoacidúria e acidose tubular renal; b) devido às manifestações clínicas dessas anomalias que em geral podem ser manejadas de maneira satisfatória, perda de glicose, formação apenas de cálculos de cistina, osteoporose, acidose metabólica, depleção de potássio. etc. ec) pela ausência de uma cistinose associada.
Síndrome de Favre-Racouchout . Síndrome congênita caracterizada por anormalidades focais do tecido conjuntivo. Manifesta-se pela presença de comedões e placas amareladas circunscritas e espessas, localizadas na face, ao redor dos olhos e nariz.
Síndrome de Felty . Síndrome da artrite reumatóide associada a esplenomegalia e distúrbios hematológicos. É observada em pacientes com artrite reumatoide de longa data e inclui esplenomegalia com leucopenia, anemia e trombocitopenia e, ocasionalmente, fenômenos vasculíticos que causam úlceras cutâneas e neuropatia periférica. O mecanismo causal dessas alterações ainda não é conhecido.
Síndrome de feminização adrenal . Variedade de síndrome adrenogenital. É manifestada no homem por ginecomastia, atrofia testicular, alterações feminoides na conformação do corpo e implantação do cabelo e aumento da excreção urinária de metabólitos estrogênicos. A causa mais comum é um tumor adrenal secretor de androstenediona, que é convertido perifericamente em estrona e estradiol ou, raramente, uma deficiência de 3-b-ol-desidrogenase, que desvia o metabolismo da androstenediona para os progestogênios.
Síndrome de feminização testicular . Síndrome de pseudo-hermafroditismo em indivíduos geneticamente masculinos. É caracterizada pela presença de testículos, feminização total da genitália externa e conformação corporal, e ausência ou hipoplasia acentuada do útero e trompas, com níveis normais de testosterona circulante. Eles são devidos a uma resistência à testosterona nos órgãos efetores.
Síndrome do álcool fetal . veja síndrome alcoólica fetal
Síndrome de Fitz-Hugh-Curtis Síndrome de perihepatite devido a diferentes infecções abdominopélvicas. Ela se manifesta por febre, dor e contratura dos músculos da parede abdominal, dor mais intensa no quadrante superior direito e, ocasionalmente, ruído de fricção auscultável ao nível do quadrante superior direito. A maior causa; comum é gonorreia; embora também tenha sido observada na salpingite por clamídia e talvez outros agentes causadores.
Síndrome da flexura esplênica . Síndrome doanginosa de origem digestiva. Inclui dor e desconforto, inespecífico no quadrante superior esquerdo do abdômen, que pode causar dor na região precordial e no ombro e braço esquerdos. Sua causa comum é o meteorismo colônico.
Síndrome de Foix . Síndrome neurológica caracterizada pelo envolvimento dos nervos cranianos V, VII, IX, X e XII, Y devido à profusão de um globo ocular. Manifesta-se por oftalmoplegia unilateral completa (nervos cranianos m, IV e VI) associada à anestesia da córnea (ramo oftálmico do nervo V), com adição de exoftalmia ipsilateral, que a diferencia da síndrome de Rochon-Duvigneau. É produzida por tumores e aneurismas do seio cavernoso ou por tumores invasivos dos seios venosos ou da sela túrcica.
Síndrome de Foix-Alajouanine . Síndrome neurológica de mielite necrosante subaguda. Manifesta-se como uma paraplegia dolorosa que é primeiro espástica e depois flácida, com atrofia muscular, arreflexia tendínea, anestesia e incontinência esfincteriana. A causa parece ser degenerativa, secundária a processos infecciosos ou de origem vascular.
Síndrome de Forbes-Albright . Síndrome endócrina devido à hipersecreção de prolactina. É caracterizada por galactorreia sustentada em mulheres que não estão no período puerperal, acompanhada de níveis elevados de prolactina circulante. É causada por tumores da adenohipófise e, menos comumente, do hipotálamo.
Síndrome de Foster-Kennedy . Síndrome de anosmia unilateral de causa orgânica. Manifesta-se apenas como anosmia unilateral e é causada por aneurismas arteriais ou, mais frequentemente, por tumores da base do lobo frontal que comprimem o bulbo e a banda olfatória.
Síndrome de Frey . Síndrome irritativa de certas condições da parótida. É caracterizada por vermelhidão, sudorese e sensação de calor no território do nervo auriculotemporal, desencadeada pela mastigação. É observada em lesões unilaterais da parótida (tumores, supurações) ou do simpático cervical.
Síndrome de Froin . Síndrome relacionada a distúrbios não infecciosos do líquido cefalorraquidiano. Define o líquido cefalorraquidiano que é xantocrômico (amarelo claro, transparente), com alto conteúdo protéico, coagulação rápida e número normal de células. Essas características são observadas nos casos de interrupção do fluxo entre os ventrículos cerebrais e as áreas mais distais.
Síndrome de galactorreia relacionada a drogas . Síndrome endócrina devido à hipersecreção de prolactina. É caracterizada por galactorreia em mulheres e por turgor mamilar ou ginecomastia em homens, que aparecem durante o tratamento com certos medicamentos (incluindo espironolactona, fenotiazina e outros tranquilizantes e antidepressivos, reserpina, metildopa, digitálicos, etc.) e que desaparecem quando seus a administração é interrompida.
Síndrome do gânglio geniculado . veja Síndrome de Ramsay-Hunt
Síndrome de Ganser Síndrome de certos distúrbios comportamentais em pacientes com histeria. Ela se manifesta em uma personalidade histérica, por amnésia, distúrbios atípicos de consciência, atos absurdos e respostas sem sentido a perguntas.
Síndrome de Garcin . Síndrome neurológica da base do crânio. Paralisia unilateral de todos os nervos cranianos, principalmente dos dez últimos, sem sinais de hipertensão intracraniana. É causada por tumores extensos ou metástases tumorais que se infiltram progressivamente na base do crânio.
Síndrome de Gardner . Síndrome hereditária caracterizada por alterações cutâneas e tendência ao desenvolvimento de processos malignos. Manifesta-se pela presença de tumores dermóides, lipomas, fibromas, osteomas e cistos epidérmicos e sebáceos, associados à polipose colônica. A incidência de adenocarcinomas do cólon é extremamente alta, a menos que o indivíduo esteja colectomizado. A imagem é transmitida como um traço autossômico dominante.
Síndrome gastrocardíaca . Síndrome cardíaca funcional de origem gástrica. Manifesta-se por extrassístoles, episódios transitórios de taquicardia ou sintomas pseudo-anginosos, não relacionados ao esforço e que costumam aparecer no período pós-prandial. Eles são atribuídos à compressão da câmara gástrica contra a parede inferior do diafragma e, com alguma frequência, há uma hérnia de hiato associada.
Síndrome de Gélineau . Síndrome de narcolepsia. Em sua forma completa, é caracterizado pela seguinte tétrade: a) ataques recorrentes e irresistíveis de sonolência diurna (narcolepsia); b) episódios breves e repentinos de perda de tônus muscular sem perda de consciência (cataplexia); c) sensação assustadora de incapacidade de mover os músculos voluntários no início do sono ou ao acordar (paralisia do sono), ed) alucinações no início do sono (hipnagógica) ou ao acordar (hipnopômpico). A causa ainda não foi esclarecida.
Síndrome de adaptação geral . Síndrome da resposta global do organismo a situações estressantes. Conjunto de reações sistêmicas consecutivas à exposição sustentada a condições de estresse (trauma, frio, calor, fadiga, etc.). É composto por três etapas: reação ao alarme, estágio de resistência e estágio de exaustão.
Síndrome de Gerstrnann . Síndrome neurológica decorrente de lesão do giro angular dominante. Combinação de agnosia tátil, dificuldade em distinguir entre direita e esquerda, agrafia, acalculia e freqüentemente apraxia de agarrar. É visto em lesões do giro angular esquerdo, entre o córtex parietal e occipital, e geralmente é de origem tumoral.
Síndrome de Gilles de la Tourette . Síndrome de tique múltiplo crônico. Eles são caracterizados por tiques faciais que aparecem na infância e progridem para gerar espasmos: amplos; estereotipados, no resto do organismo, e que são tipicamente acompanhados por ecolalia e coprolalia. A causa é atribuída a uma combinação de fatores genéticos e ambientais, e o distúrbio persiste por toda a vida.
Síndrome de Goodpasture . Síndrome associada a glomerulonefrite e distúrbios pulmonares consumo de hemop . O ponto de partida é uma condição respiratória com hemoptise e infiltrados pulmonares bilaterais difusos, seguidos por anemia e glomerulonefrite rapidamente progressiva manifestada por hematúria, proteinúria grave, hipertensão arterial e insuficiência renal. O resultado geralmente é fatal e é atribuído a fenômenos anti-imunidade.
Síndrome de Gorlin-Goltz . Síndrome hereditária caracterizada por alterações cutâneas e tendência ao desenvolvimento de processos malignos. É definida pela presença de múltiplos carcinomas basocelulares, cistos epidermóides e covinhas nas palmas das mãos e plantas dos pés; além de fibromas ovarianos e cistos maxilares e, em certa proporção dos casos, hipertelorismo e alterações metacarpais. É transmitido como um traço autossômico dominante e foi demonstrada uma tendência definida para o desenvolvimento de fibrossarcomas da mandíbula e meduloblastomas.
Síndrome de Gougerot-Carteaud . Síndrome da papilomatose confluente e reticulada. É caracterizada pela presença de um grande número de pápulas discretas; que então convergem para se tornarem pápulas verrucosas. Ele está localizado na linha média do tronco e nas dobras de flexão do cotovelo e regrede lentamente. Geralmente afeta meninas em idade puberal.
Síndrome de Gradenlgo . Síndrome neurológica com envolvimento dos nervos cranianos V e VI. É definida pela presença de paralisia unilateral do olhar externo (lesão do sexto nervo craniano) acompanhada de cefaleia e neuralgia facial ipsilateral (lesão do quinto nervo craniano). É observada em processos supurativos da orelha, média e em tumores da fossa escarpada.
Síndrome de Groenblad-Straudberg . Síndrome de pseudoxantoma elástico. Suas características distintivas são: a) na pele, o aparecimento de áreas romboidais degenerativas, atrofia e flacidez, que predominam no pescoço, axilas e virilhas e se desenvolvem após 30-40 anos; b) diminuição progressiva da visão, relacionada à presença de bandas angióides, de coloração marrom ou marrom-acinzentada na superfície do retículo ec) sinais crescentes de insuficiência circulatória periférica produzida por calcificação e oclusão de artérias de médio calibre, incluindo os do membro superior. A condição é de causa desconhecida e é transmitida como um traço autossômico recessivo.
Síndrome de Guillaln-Barré . Síndrome da polineurite idiopática aguda: enfraquecimento rapidamente progressivo dos neurônios motores ascendentes que geralmente ocorre após uma infecção respiratória ou entérica. Começa com parestesias dos pés, seguidas de paralisia flácida e fraqueza das pernas; que sobem para os braços, tronco e rosto; é acompanhada por febre baixa, paralisia bulbar, ausência ou diminuição dos reflexos do tendão e aumento das proteínas do líquido cefalorraquidiano sem crescimento celular concomitante. A causa é considerada imunológica e a recuperação é completa em 75% dos pacientes.
|
|
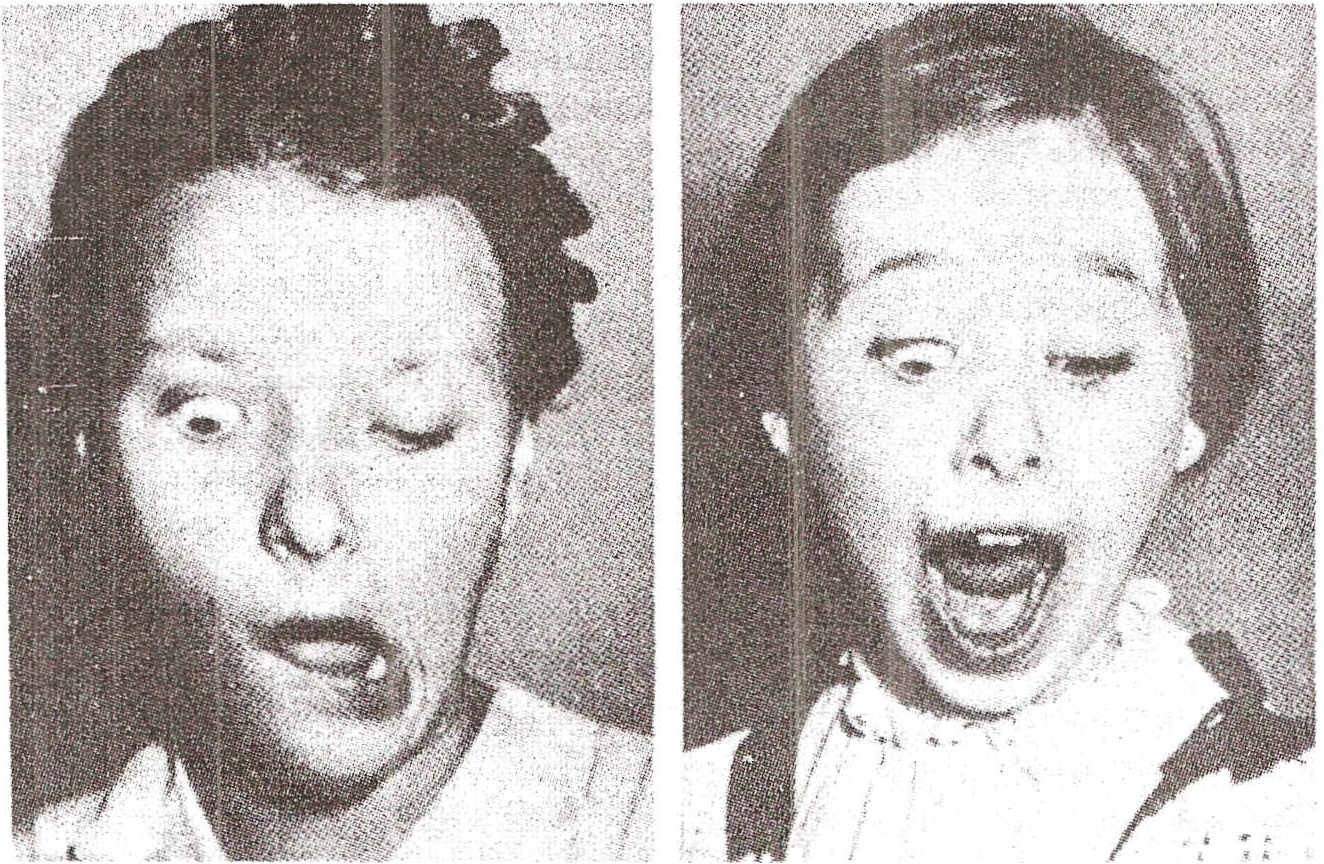
Síndrome de Gunn . Síndrome relacionada aos movimentos associados de uma pálpebra e da mandíbula. É caracterizada por ptose palpebral unilateral, quase sempre congênita, e elevação exagerada da pálpebra ptosa ao movimentar a mandíbula no sentido contralateral; Em alguns casos, o inverso também ocorre: a estimulação da córnea causa movimentos da mandíbula para o lado oposto. A causa é desconhecida e não há outros distúrbios corporais.
Síndrome de Hallopeau-Siemens . Síndrome da epidermólise bolhosa polidisplásica. É constituído por bolhas que se desenvolvem desde cedo na vida e que cicatrizam, deixando cicatrizes, nas quais podem aparecer epiteliomas. As membranas mucosas são freqüentemente afetadas. É uma doença hereditária recessiva.
Síndrome de hamartoma múltiplo . Síndrome hereditária caracterizada por alterações cutâneas e tendência ao desenvolvimento de processos malignos. Manifesta-se pela presença de: a) pápulas verrucosas na pele; b) fibromas em mucosa oral; c) múltiplos hamartomas em outras regiões do corpo (fibrocistose mamária, neuromas, adenomas de tireoide, lipomas e hemangiomas), ed) tendência aumentada para desenvolver carcinomas. glândula mamária e tireóide. O distúrbio é transmitido como um traço autossômico dominante.
Síndrome de Hamman-Rich . Síndrome de fibrose pulmonar idiopática. É caracterizada pelo desenvolvimento de tecido fibroso no interstício pulmonar, de evolução crônica, subaguda ou aguda. Há dispneia progressiva e, posteriormente, permanente, baqueteamento digital e alteração no transporte de gases pela parede alveolar. É irreversível e de causa desconhecida.
Síndrome de Harris . Síndrome de hiperinsulinismo endógeno. Manifesta-se por episódios mais ou menos frequentes de inquietação, palidez, sudorese, taquicardia, confusão mental e distúrbios da visão, relacionados com uma diminuição acentuada dos níveis de glicose plasmática. É produzida por hipersecreção de insulina de tumores ou hiperplasia difusa das células beta das ilhotas de Langerhans.
Síndrome de Hedblom . Inflamação aguda da síndrome do diafragma. Manifesta-se por dor inspiratória, imobilidade da parte inferior do tórax durante a inspiração e dor bilateral na parte superior do abdome, sem patologia abdominal que justifique o quadro. É devido a uma miosite aguda primária do diafragma.
Síndrome de Heerfordt . Síndrome da febre uveoparótida. É caracterizada por inflamação uveal e edema da parótida, associada à paralisia facial bilateral (diplegia facial) com ou sem paralisia de outros nervos cranianos. É uma das variantes de iniciação da sarcoidose.
Síndrome de Heidenhain . Síndrome neurológica de demência pré-senil. É uma doença de evolução rápida acompanhada de cegueira cortical, disartria, ataxia, atetose e rigidez generalizada, sem alterações no fundo do olho. O distúrbio é degenerativo e de causa desconhecida.
Síndrome de hemangioma com trombocitopenia . veja a síndrome de Kasabach-Merritt .
Síndrome do hematoma subdural . Síndrome neurológica de coleções venosas intracranianas. É manifestada por cefaleia, sonolência, confusão mental, sinais motores focais (monoparesia, hemiparesia), assimetria pupilar, papiledema e, eventualmente, estupor, coma e convulsões cônicas. É geralmente de progressão insidiosa, sobreposta e produzida pela lenta expansão de um hematoma intracraniano de origem venosa, em pacientes com ou sem história marcante de traumatismo cranioencefálico, de curta ou média duração (de alguns dias a um ou mais meses)
Síndrome de hemianopia bitemporal . Síndrome neuro-oftalmológica de lesão bilateral do nervo óptico. O quadro é causado por adenomas da hipófise, meningiomas da sela túrcica ou aneurismas saculares do polígono de Willis, que ao se expandir comprimem simultaneamente a porção interna de ambos os nervos ópticos, ou seja, as fibras que vão para a metade nasal de ambas as retinas. Como consequência, há cegueira para objetos localizados no lado temporal de ambos os campos visuais (hemianopia bitemporal).
Síndrome da fenda esfenoidal . veja síndrome de Rochon-Divigneau.
Síndrome de hiperabdução . veja Síndrome do tórax estreito superior .
Síndrome de hipercalemia . Síndrome muscular e eletrocardiográfica de hipercalemia. Torna-se evidente quando os níveis de potássio sérico excedem 6 mEq / L e é caracterizado por fraqueza muscular crescente, hiporreflexia ou arreflexia osteotendinosa e alterações típicas no eletrocardiograma: primeiro, ondas T agudas aparecem em todas as derivações e, em seguida, sucessivamente, alongamento PR, desaparecimento da onda P, alterações no complexo QRS e, eventualmente, fibrilação ventricular e parada cardíaca. É observada na doença de Addison, nos estágios terminais da insuficiência renal crônica e principalmente nas formas. hipercatabólica de insuficiência renal aguda.
Síndrome de hiperceratose palmoplantar . Síndrome hereditária com alterações cutâneas e tendência ao desenvolvimento de processos malignos. É caracterizada pela presença de hiperceratose das palmas das mãos e da planta dos pés, que se inicia já na segunda infância, sem outras anormalidades: detectáveis. A incidência subsequente de carcinomas de esôfago é extremamente alta e a doença é transmitida como traço autossômico dominante.
Síndrome hipercinética da infância . Síndrome de aceleração das funções psicomotoras na criança. É caracterizada por hiperatividade motora, impulsividade, inquietação, acentuada incapacidade de fixar a atenção e baixa tolerância à frustração. A causa não é conhecida, embora na maioria dos casos não seja orgânica, e a condição geralmente desaparece na adolescência.
Síndrome de hipersonia com apneia noturna . Síndrome apnéica associada à obesidade de diferentes etiologias. O quadro típico é de uma hipersonia diurna com pausas; de frequência e duração anormais que ocorrem durante o sono. É observada na síndrome de Pickwick, obesidade simples, mixedema e outros estados de doença.
Síndrome de hipertensão portal . Síndrome de hipertensão venosa devido à obstrução do sistema porta. É manifestada por ascite e edema periférico, esplenomegalia congestiva, desenvolvimento de circulação venosa colateral (abdome e parede torácica anterior), dilatação de outros sistemas venosos (varizes esofágicas), oligúria e eventualmente hemorragias digestivas (varizes esofágicas rompidas). É vista como uma manifestação primária na pilephlebite da veia porta e, secundariamente, na cirrose hepática e outros processos obstrutivos.
Síndrome de hiperventilação . Síndrome de hiperventilação pulmonar não orgânica. É uma imagem de angústia e sensação de falta de ar, dormência e parestesia no rosto e nos membros, e cãibras nas mãos e nos pés. Geralmente ocorre durante a noite, quando o indivíduo tenta adormecer ou logo após adormecer. A causa é a hiperventilação desencadeada por fatores emocionais.
Síndrome de hipocalemia . Síndrome muscular e eletrocardiográfica de hipocalemia. É caracterizada por fraqueza geral que pode até levar à paralisia completa. (como visto na paralisia periódica familiar), acompanhada por anormalidades eletrocardiográficas típicas: achatamento generalizado das ondas: T, ondas U proeminentes, rebaixamento de ST e, eventualmente, parada cardíaca em assistolia. A causa mais frequente é a ingestão de diuréticos, por automedicação ou por prescrição médica e também pode ser causada pelo abuso crônico de laxantes, nas fotos de vômitos profusos ou diarreia abundante, etc.
Síndrome de hipotensão intracraniana . Síndrome neurológica de hipotensão do líquido cefalorraquidiano. Manifesta-se por náusea, vertigem e zumbido, bradicardia, cefaleia que alivia em decúbito, rigidez de nuca e papiledema bilateral leve a moderado. É uma complicação relativamente frequente de traumatismo cranioencefálico e deve-se à interrupção da formação do líquido cefalorraquidiano com a consequente tendência ao colapso dos espaços ventriculares.
Síndrome de hipotensão ortostática idiopática . Síndrome associada à degeneração periférica ou central de estruturas do sistema nervoso autônomo. É caracterizada por hipotensão ortostática cuja gravidade pode causar síncope ou convulsões. A condição é progressiva, é observada em idosos e é acompanhada por anidrose ascendente, diminuição do metabolismo basal e produção de norepinefrina, laceração e secreção de saliva deficientes, atonia da bexiga e outras manifestações.
Síndrome de hipoventilação idiopática . Síndrome de hipoventilação com manifestações clínicas, corrigível voluntariamente e de causa desconhecida. É uma condição rara que acomete indivíduos sem patologia pulmonar ou da parede torácica, sem alterações nos testes de função respiratória e sem alterações detectáveis no sistema nervoso central. É caracterizado: a) por uma tendência involuntária e insensível sustentada de hipoventilar, suficientemente importante para que o PC02 aumente e a P02 sanguínea diminua, e para o indivíduo estar em um estado de fraqueza permanente, cansaço e sonolência, e b) pela capacidade do indivíduo de reverter a situação na medida em que está ciente do que está acontecendo com ele e intensifica voluntariamente a atividade ventilatória.Os valores de hematócrito são superiores a 50% e episódios respiratórios periódicos são observados durante o sono na maioria dos casos.
Síndrome de Hippel-Landau . Síndrome neuro-oftalmológica associada a malformações vasculares congênitas. Anatomicamente, é definida pela presença simultânea de múltiplos hemangiomas capilares na retina e um ou mais hemangioblastomas no cerebelo, de crescimento lento e com formações císticas. E clinicamente, devido à dor de cabeça e papiledema, e manifestações progressivas de ataxia cerebelar e perda de visão. Existem casos de aparecimento espontâneo e outros que são transmitidos como traço autossômico dominante.
Síndrome de Holt-Oram . Síndrome associada a malformações do sistema cardiovascular e esquelético. Associação de defeitos do septo interatrial, ou outras anomalias cardíacas, com hipoplasia das clavículas e malformações dos membros superiores.
Síndrome de Holzknecht-Jacobson . Síndrome radiológica de atelectasia pulmonar. É caracterizada por opacidade do campo pulmonar afetado, desvio do mediastino para esse lado; elevação do diafragma ipsilateral e retração dos espaços intercostais.
Síndrome do homem rígido . veja Síndrome de Wolt Man-Moersch
Síndrome do ombro-mão . veja Síndrome da Distrofia Simpática Reflexa .
Síndrome de Homen . Síndrome neurológica da degeneração do núcleo lenticular. É uma condição progressiva e irreversível caracterizada por vertigem, disartria, ataxia, rigidez que predomina nos membros inferiores e demência. A origem é genética.
Síndrome de Horner . veja Síndrome de Claude Bernard Horner .
Síndrome de Horton . Síndrome da cefaleia histamínica É caracterizada por ataques unilaterais de dor nos olhos e na testa, com início e término súbitos; há vermelhidão, calor e suor no lado afetado devido à dilatação da artéria temporal. Ela predomina em homens, durante o sono, e é atribuída a uma descarga local de histamina
Síndrome de Hunt . Síndrome da paralisia agitadora juvenil. Manifesta-se desde os primeiros anos de vida por hipertonia muscular e características faciais e atitude geral típica da doença de Parkinson. É familiar e se deve à degeneração progressiva do globo pálido e de outros núcleos vizinhos.
Síndrome de Hunter . Síndrome de mucopolissacaridose II. É caracterizada por: a) escafocefalia com pescoço curto, sobrancelhas hirsutas, língua grande e proeminência lateral das paredes externas das órbitas; b) rigidez articular, com pés ocos e mãos em flexão; c) ausência de opacidades corneanas ed) surdez, mas com retardo mental menos grave que o da síndrome de Hurler. É observada apenas no sexo masculino, que pode chegar à meia-idade, e se deve à deficiência da iduronato sulfatase, transmitida como traço recessivo ligado ao X.
Síndrome de Hurler . Síndrome de mucopolissacaridose 1H. Suas características básicas são: a) cabeça volumosa com escafocefalia, sobrancelhas hirsutas; testa e olhos proeminentes, língua grossa e bochechas salientes (gárgulas, devido à sua semelhança com gárgulas na arquitetura gótica); b) baixa estatura, pescoço e membros, com cifose acentuada e ventre proeminente; c) opacidades da córnea ed) surdez e retardo mental severo. O distúrbio é genético, devido a uma deficiência de aL-iduronidase e a morte geralmente ocorre antes dos 10 anos de idade
Síndrome de incontinência pigmentar . veja síndrome de Dloch-Sijlzberger .
Síndrome de imunodeficiência combinada grave . Síndrome congênita relacionada a uma insuficiência global dos mecanismos imunológicos. É caracterizada por graves distúrbios da imunidade humoral e celular, com deficiência de imunoglobulina e linfopenia acentuada, incluindo células T e B. Existem infecções repetidas e os pacientes geralmente morrem no primeiro ano de vida. O distúrbio é do tipo hereditário congênito ou se apresenta na forma de casos esporádicos.
Síndrome de imunodeficiência com timoma . Síndrome de imunodeficiência de causa indeterminada É observada em pacientes com timomas de células fusiformes e é caracterizada pela ausência ou diminuição acentuada de linfócitos B na circulação e de seus precursores na medula óssea, juntamente com eosinopenia, baixos níveis de imunoglobulinas plasmáticas e ocasionalmente, aplasia eritroide.
Síndrome da insuficiência poliglandular . Síndrome de insuficiência simultânea de dois ou mais sistemas endócrinos. A síndrome é produzida por diferentes quadros de hipofunção endócrina (insuficiência adrenal, hipoparatireoidismo, tireoidite linfocítica, insuficiência gonadal) e pelas manifestações que resultam da combinação de dois ou mais deles entre si, aos quais diabetes e diabetes são frequentemente associados a nanismo. Nenhuma anormalidade significativa é detectada na adenohipófise e suas características básicas são os baixos níveis de hormônios circulantes e a existência de anticorpos contra dois ou mais sistemas endócrinos.
Síndrome da dificuldade respiratória do recém-nascido . Síndrome do desconforto respiratório agudo de etiologia múltipla. Seus dois sinais principais são dispneia e taquipneia acentuada (mais de 60 respirações por minuto), acompanhada de graus variáveis de taquicardia, cianose e alterações do estado ácido-básico. É produzida por causas: central (depressão medicamentosa, imaturidade cerebral ou anoxia, etc.) ou periférica: processos infecciosos, aspiração de líquido amniótico, atelectasia, imaturidade pulmonar (síndrome de Wilson-Mikity), depósito de material hialino nos alvéolos e bronquíolos (síndrome da membrana hialina), etc.
Síndrome interolivar . veja síndrome de Dejerine .
Síndrome de intolerância à frutose . veja Síndrome de Deficiência Hepática de Aldolase .
Síndrome de intoxicação digital . Síndrome produzida pela ingestão de doses excessivas de digitálicos. Inclui distúrbios digestivos, que geralmente são os primeiros a aparecer (anorexia acentuada, estado de náusea persistente), distúrbios visuais (visão turva ou colorida, escotomas) e anormalidades eletrocardiográficas cuja gravidade é geralmente proporcional à intensidade da intoxicação, arritmias do tipo aparecer. Extrassístole isolada, bigeminia extra-sistólica ou extra-sístole ventricular multifocal, seguida por bloqueio atrioventricular de primeiro ou segundo grau e, eventualmente, bloqueio completo.
Síndrome de radiação . Síndrome produzida por superexposição aguda aos raios roentgen. Manifesta-se por anorexia, fraqueza e febre, sede e prostração intensas, seguidas após pouco tempo por anemia, granulocitopenia, emagrecimento, queda de cabelo e alterações generalizadas que em casos de irradiação maciça (vazamentos radioativos, explosões nucleares, etc.) levar rapidamente à morte do indivíduo.
Síndrome de Ivemark . Síndrome de agenesia do baço com anormalidades cardíacas e viscerais. É caracterizada por agenesia do baço, malformações cardíacas: geralmente com shunt da direita para a esquerda e inversão das vísceras de grau variável. O quadro é congênito e deve-se a alterações do desenvolvimento que ocorrem por volta da quarta ou quinta semana de vida intrauterina.
 Síndrome de Jaccoud Síndrome de Jaccoud
|
Síndrome de Jaccoud . Sim ND r ou m e um poliartr i t i s c Ró nica pró g resi é d ee v ou lu c ç ã o n para lhe pica .Ele define uma poliartrite crônica progressiva com consequências praticamente comparáveis às da artrite reumatóide (fibrose periarticular, rigidez, desvio ulnar da mão, etc.), mas que não começa como tal, mas sim como uma variante evolutiva em um indivíduo que era um portador, inicialmente, das manifestações reversíveis típicas do reumatismo poliarticular agudo (febre reumática).
Síndrome de Jackson . Sim n d ro m e neurol ou p or rl e são lógica do tronco c e rebra l c ou n co mprom iso de n e va i ou cranial I X e X se manifesta por paralisia de metade da língua do lado da lesão (nervo IX) mais os sinais da síndrome de Avellis. É causada por tumores ou focos de amolecimento ao nível do teto do bulbo e do trato corticoespinhal.
Síndrome de Jacod . S iNDr ou m e ne urooftálmico po rl e se ou n nervo cran e para o é. É unilateral e se caracteriza por neuralgia do território trigêmeo, cegueira e oftalmoplegia completa. É produzida por tumores nasofaríngeos que se estendem até a junção petroesfenoidal e afetam os nervos cranianos II, III, V e VI.
Síndrome de Jaddasohn-Lewandowsky . Sind r ou m e d e o paquioníquia cong ê n i ta. É caracterizada pelo espessamento das unhas; onicogripose, hiperceratose dos joelhos, cotovelos, palmas das mãos e plantas dos pés, leucoplasia oral e hiperidrose das mãos e pés. É herdado como um traço autossômico dominante.
Síndrome de Jackson-Wartenhorst . Sim Não drome d e l um p ou licondritis recidi v ante. É um processo inflamatório doloroso e febril da cartilagem das orelhas e do nariz, que leva à atrofia e pode afetar a cartilagem do trato respiratório e as costelas. É acompanhada por artrite, episclerite, irite, miocardite e insuficiência aórtica. A causa é desconhecida.
Síndrome de Jervell e Lange-Nielsen . veja síndrome QT .
Síndrome de Kartagener . S i n d Roma d e um Cua d r ou m para lformacione s cardi ov uma s c ular é e d e l como vias resp ir para a r ias . É definida pela presença de dextrocardia, situs inversus e bronquiectasia e pela tendência a desenvolver infecções pulmonares e sinusais recorrentes. O distúrbio é herdado e transmitido como traço autossômico dominante.
Síndrome de Kasabach-Merritt . Sim n dromo trombocitopênico relacionado à presença de hemangioma, Manifesta-se por trombocitopenia acentuada em pacientes com hemangioma gigante, sem alterações significativas na medula óssea, exceto por excesso de megacariócitos. O quadro é típico do recém-nascido e se normaliza com a retirada do hemangioma.
Síndrome de Kennedy . Síndrome n e urooftalmoló g i c ou ou r i g e n tum ou ral. É caracterizada por cegueira unilateral e papiledema no lado oposto. É causada por tumores do lobo frontal, ou meningiomas da crista esfenoidal, que progressivamente comprimem um nervo óptico (cegueira ipsilateral) e obstruem o retorno venoso do lado oposto (papiledema contralateral).
Síndrome de Kimmelstiel-Wilson . Síndrome de d i a b et e s s c Arina a s ou c iado de hipertensão ou nl e se ou n é r em que o e s d e g e n e rativa s . As últimas consistem em glomerulo-hialinose intercapilar e se manifestam por hematúria, albuminúria e cilindrúria, uma síndrome nefrótica e insuficiência renal progressiva.
Síndrome de Kinsbourne . Síndrome de e pil e psi de m io c lon ic como o infa n cia . É caracterizada por contrações mioclônicas do tronco, extremidades e face, com ataxia dinâmica e tremor intencional. A causa é desconhecida, embora em 50% dos casos haja um neuroblastoma oculto.
Síndrome de Kleine-Levin : sind r ome hyper s ou m n i, por exemplo, river dica. É caracterizada por períodos de hipersonolência que duram de dias a semanas, alternando com períodos assintomáticos de meses a anos. Na primeira, o indivíduo apresenta apetite voraz e pode manifestar alterações de humor e comportamento sexual (hiperatividade, exibicionismo) juntamente com alucinações, desorientação e distúrbios de memória. Geralmente começa na juventude e a causa é desconhecida.
Síndrome de Klinefelter . Síndrome de c ause generi ca c um r um c te ri z de fazer por perturbações do sistema endócrino e um ou m um l ed s GE e t um l e s. Distingue-se pela aparência eunucóide ou fracamente masculinizada do paciente, ginecomastia, testículos pequenos e hipoplásicos, hialinização dos túbulos seminíferos com espermatogênese e preservação das células de Leydig, níveis elevados de gonadotrofinas urinárias (FSH) e diminuição da secreção de androgênios.O sexo genético é geralmente feminino, há três ou mais conjuntos de cromossomos sexuais (XXY, XXXY ou XXYY, XXXXY) e os cromossomos autossômicos são normais.
Síndrome de Klippel-Feil . Síndrome produzida pela ausência congênita de vértebras cervicais. Suas características básicas são: a) encurtamento acentuado ou ausência virtual do pescoço, de forma que a cabeça parece repousar diretamente na parte superior do tronco; b) incapacidade de virar a cabeça; c) implantação extremamente baixa dos pelos da nuca ed) as radiografias mostram diminuição do número de vértebras cervicais, consolidação das vértebras existentes em um único bloco e abertura dos arcos vertebrais posteriores (espinha bífida).
Síndrome de Klippel-Trenaunay-Weber . Síndrome associada a malformações unilaterais de origem congênita. O quadro geralmente afeta um membro e é caracterizado pelo crescimento exagerado do membro devido à hipertrofia óssea e de tecidos moles, muitas vezes com sindactilia ou outras malformações, e pela coexistência de anomalias vasculares como hemangiomas cutâneos, dilatações de veias varicosas ou nevos em chamas. A causa ainda não foi estabelecida.
Síndrome de Konig . Síndrome de quadro de dor intermitente em fossa ilíaca direita com alteração do trânsito intestinal. É caracterizada pelo aparecimento intermitente de manifestações ao nível da fossa ilíaca direita (cólicas e distensão localizada, seguidas de roncos que aliviam rapidamente os sintomas) e por períodos de constipação que se alternam com outros de diarreia. É observada em processos inflamatórios ou neoplásicos que afetam o ceco ou áreas vizinhas.
Síndrome de Korsakoff . Síndrome de psicose amnésica confabulatória. É observada em alcoolistas crônicos e é caracterizada por transtornos de orientação, suscetibilidade a estímulos e sugestões externas, alucinações, confabulação e principalmente amnésia, que é desproporcional às demais manifestações mentais e comportamentais. Geralmente ocorrem alterações polineuríticas (mão pendular, etc.).
Síndrome de Koshevnicoff . Síndrome de certas formas graves de epilepsia motora. Inclui crises convulsivas de repetição, hipertemia, delírio, perda de consciência e paralisia de distribuição variável, juntamente com prostração e persistência dos movimentos mioclônicos nos períodos intercríticos.
Síndrome de Kwashiorkor . Síndrome de desnutrição protéica calórica severa na infância. É observada em crianças pequenas e é caracterizada por acentuada anorexia, edema e hipoalbuminemia, alterações na pele (despigmentação com manchas eritematosas, dermatoses, várias), descoloração nas faixas de cabelo, aparência ruim e falta de resposta emocional. É devido a deficiências, dietas múltiplas e prolongadas.
Síndrome da lágrima de crocodilo . veja síndrome de Bogorad .
Síndrome de Landry-Guillain-Barré . veja síndrome de Guillain-Barre .
Síndrome de Laubry-Soule . Síndrome digestiva associada a infarto do miocárdio. É observada em alguns pacientes com infarto do miocárdio e se manifesta por meteorismo localizado ao nível do canto esquerdo do cólon e da câmara gástrica. É considerado um fenômeno reflexo.
Síndrome de Launois . Síndrome que acompanha certas formas de hiperpituitarismo. É um quadro de gigantismo que se inicia antes do fechamento das epífises, geralmente na puberdade, e que resulta de uma hipersecreção de hormônios hipofisários de origem tumoral (adenomas eosinofílicos e, às vezes, adenomas cromofóbicos).
Síndrome de Laurence-Moon-Biedl . Síndrome hereditária produzida por disfunção hipotalâmica. Seus componentes são os da síndrome adiposogenital de Frohlich (obesidade feminoide, infantilismo sexual, retardo mental, etc.), aos quais se somam retinite pigmentosa e polidactilia. Não há alterações na adenohipófise e a condição é de origem genética.
Síndrome do leite-álcali . Síndrome de hipercalcemia devido à ingestão prolongada de álcalis absorvíveis. Suas manifestações incluem prurido, fraqueza muscular e dor reumatiforme, polidipsia e poliúria, hipercalcemia sem hipercalciúria e calcinose generalizada que pode eventualmente levar à insuficiência renal. O quadro é típico de úlceras que consomem grandes quantidades de leite e álcalis, e sua incidência diminuiu notavelmente desde a introdução de antiácidos não absorvíveis.
Síndrome de Leriche . Síndrome de obstrução crônica da porção terminal da aorta abdominal. É caracterizada por palidez e resfriamento dos membros inferiores, fraqueza dos músculos do quadril, pernas ou tornozelos durante a caminhada ou exercício, impotência sexual e ausência de pulsos femorais.
Síndrome de Lermoyez . Síndrome de certas alterações transitórias da função auditiva e vestibular. Inclui zumbido e surdez de início súbito, que precedem um ataque de vertigem e diminuem quando a vertigem se instala. É atribuído a espasmos sustentados da artéria auditiva interna.
Síndrome de Lewis . Síndrome hereditária devido a anormalidades congênitas no metabolismo de carboidratos. Inclui retardo mental de grau variável, distúrbios neurológicos (principalmente convulsões) e convulsões hipoglicêmicas que predominam à noite. A causa é uma deficiência congênita da glicogensintetase hepática.
Síndrome de Lichtheim . Síndrome de degeneração combinada subaguda da medula espinhal. É manifestado por parestesias; ataxia, marcha instável e, posteriormente, paraplegia espástica. É visto na anemia idiopática ou perniciosa de Biermer e é devido à degeneração das colunas lateral e posterior da medula espinhal
Síndrome de Liddle . Síndrome de pseudo-hiperaldoseronismo. É caracterizada por hipertensão e alcalose hipoclorêmica, com secreção insignificante de aldosterona. É hereditário, provavelmente autossômico dominante, e a causa é desconhecida.
Síndrome de Lightwood . Síndrome da incapacidade dos rins de excretar urina suficientemente ácida. Ela se manifesta como acidose metabólica persistente com hipercloremia e, eventualmente, com depleção de potássio e hipercalciúria. É devido a um defeito primário na função dos túbulos renais (de tipo familiar ou de aparência esporádica) ou a uma disfunção secundária a estados de disproteinemia, hipervitaminose D, hipertireoidismo, etc.
Síndrome linfogranular mucocutânea . Síndrome de dermatite confluente da infância. Inicia-se como quadro febril agudo com poliadenopatia, ao qual se acrescenta erupção maculoeritematosa que se caracteriza por ser confluente e edemaciada, predominantemente nas mãos e pés, e pela descamação abundante que acompanha a resolução das lesões. Foi observado pela primeira vez no Japão e sua causa é desconhecida.
Síndrome de Loffler . Síndrome pulmonar com manifestações alérgicas difusas. É caracterizada por asma dispneia com tosse grave e rebelde, leucocitose leve ou moderada com eosinofilia acentuada e a presença de múltiplos granulomas pulmonares que se manifestam como infiltrados difusos e fugazes nas radiografias de tórax. A etiologia da doença ainda não é conhecida com precisão.
Síndrome de Louis-Bar . Síndrome de ataxia-telangiectasia. É caracterizada por ataxia cerebelar progressiva, telangiectasia oculocutânea, infecções frequentes dos seios da face e dos pulmões e movimentos anormais dos olhos. Geralmente existe uma deficiência imunológica. É uma condição hereditária transmitida como um traço autossômico recessivo.
Síndrome de Lown-Ganong-Levine . Síndrome eletrocardiográfica e clínica de excitação atrioventricular anormal. As manifestações clínicas são as mesmas da síndrome de Wolff-Parkinson-White, mas, ao contrário disso, a única anormalidade eletrocardiográfica é o encurtamento do PR.
Síndrome de Lubarsch-Pick . Síndrome congênita produzida por depósito de substância amilóide. É caracterizada pela deposição de amiloide atípico materno na pele e tecido muscular, com esclerodermia e macroglossia. A causa é desconhecida.
Síndrome de Lutembacher . Síndrome associada a malformações cardíacas congênitas. Designa a combinação de defeito do septo atrial (devido à persistência do forame oval), estenose mitral e hipertrofia secundária do átrio esquerdo.
Síndrome de Lyell . Síndrome de necrólise epidérmica tóxica. Consiste em dermatite esfoliativa severa, precedida por eritema e grandes bolhas. Geralmente é infeccioso (estafilococos) na infância ou tóxico (devido aos medicamentos) em adultos.
Síndrome alveolar completa . Síndrome radiológica da ocupação do espaço alveolar. É caracterizada pela presença de imagens nodulares difusas moderadamente densas, com limites difusos, que tendem a convergir em napa e envolver um lobo ou segmento pulmonar, ou que se distribuem uniformemente em ambos os campos pulmonares. É observada na broncopneumonia ou na fase inicial da pneumonia, câncer alveolar, edema agudo do pulmão e assim por diante.
Síndrome de macrogenitosomia precoce . veja Síndrome Pineal .
Síndrome de Mallory-Weiss . Síndrome da ruptura esofágica devido ao estresse. É um quadro de hematêmese, frequentemente maciça, que ocorre em pacientes que apresentaram vômitos repetidos e persistentes. Ela ocorre a partir de lacerações longitudinais nas proximidades da junção esofagogástrica.
Síndrome de piscadela . veja síndrome de Gunn .
Síndrome de Marcus Gunn. veja síndrome de Gunn .
Síndrome de Marchiafava-Micheli . Síndrome de hemoglobinúria paroxística noturna. É caracterizada por episódios de hemólise paroxística com hemoglobinúria, geralmente noturna, aumento da hemoglobina plasmática e, às vezes, leucopenia e trombocitopenia. É prevalente em adultos jovens e se deve a uma mutação nas células-tronco da medula óssea, com sensibilidade exagerada ao complemento.
Síndrome de Marfan . Síndrome produzida por anomalias congênitas dos elementos fibrosos do tecido conjuntivo. Normalmente se manifesta por: a) estatura alta, magreza que às vezes é extrema e tórax de conformação anormal; b) membros graciosos e desproporcionalmente longos em relação ao tronco, o que é particularmente evidente nos segmentos mais distais (aranodactilia); c) escafocefalia com palato ogival e protusão do acetábulo; d) anormalidades oculares, incluindo ectopia do cristalino, opacidades da córnea e, ocasionalmente, miopia e glaucoma;e) fraqueza muscular, hipermobilidade das articulações e diferentes deformações ósseas; e f) tendência acentuada ao desenvolvimento de insuficiência aórtica ou aneurismas aórticos, em decorrência da fraqueza congênita da camada média deste vaso. É uma imagem genética transmitida como um traço autossômico dominante.
Síndrome de Marie-Bamberger . Síndrome de osteoartropatia pneumática hipertrófica. É secundária à doença pulmonar crônica e é caracterizada por hiperemia e edema do periósteo, com neoformação da matriz óssea e posterior calcificação. Isso produz um espessamento simétrico nos punhos e tornozelos, metacarpos e metatarsos, e nas extremidades dos ossos longos, geralmente há “dedos em coxa”. O mecanismo causal ainda não foi esclarecido.
Síndrome de Maroteaux-Lamy . Síndrome de mucopolissacaridose VI. Suas características básicas são: a) fácies com feições grosseiras, porém menos acentuadas que na síndrome de Hurler, com estatura, pescoço e tronco curtos; b) hepatoesplenomegalia e tendência ao desenvolvimento de hérnias ec) ausência de retardo mental e desenvolvimento lento e tardio de opacidades corneanas. A condição, que também tem uma forma leve, é causada por uma deficiência de arilsulfatase B.
Síndrome de Mayer-Rokitansky-Kuster . Síndrome de amenorréia primária de origem genética. É caracterizada pela ausência ou hipoplasia acentuada da vagina, um útero rudimentar às vezes associado a anormalidades do sistema urinário e ocasionalmente a anormalidades esqueléticas. Tanto as características sexuais secundárias quanto a função ovariana são normais e, se o útero rudimentar tiver endométrio, pode até haver sangramento menstrual que, quando retido, produz dor abdominal de aparência cíclica.
Síndrome de Meigs . Síndrome associada à presença de tumores ovarianos. É a combinação de ascite e hidrotórax com um tumor ovariano, geralmente um fibroma.
Síndrome de Melkersson-Rosenthal . Síndrome de paralisia facial com edema crônico dos tecidos moles. É uma paralisia facial recorrente associada a edema facial crônico e persistente, que predomina nos lábios e na língua. A causa não foi esclarecida.
Síndrome de Ménétrier . Síndrome de obstrução do ducto torácico. Edema duro, acometendo membros inferiores, abdome inferior e hemitórax e braço esquerdo, associado a hidrotórax ou quilotórax e derrame peritoneal. A causa geralmente é tumor.
Síndrome de Méniere . Síndrome de vertigem recorrente, acompanhada de zumbido e surdez. Geralmente começa por volta dos 50 anos de idade e se manifesta por ataques agudos de vertigem, surdez geralmente unilateral e zumbido no ouvido afetado. A forma clássica é decorrente de hemorragia labiríntica, enquanto em outros casos resulta de processos degenerativos na orelha interna.
Síndrome meníngea . Síndrome neurológica resultante da irritação das membranas meníngeas. O quadro é típico e se manifesta por: a) fotofobia e cefaleia intensa, persistente e rebelde a qualquer analgésico; b) vômitos com fluxo central fácil e constipação persistente; c) rigidez em extensão de tronco e pescoço, com hiperestesia cutânea e hiperreflexia muscular; d) distúrbios de consciência, convulsões, coma, e) alterações quantitativas e qualitativas do líquido cefalorraquidiano (hipertensão, pleocitose, presença de sangue ou material purulento, etc.). É produzido por hemorragias, infecções e processos irritativos.
Síndrome de miastenia gravis . Síndrome de fraqueza muscular relacionada a alterações da junção neuromuscular que impedem a transmissão normal dos impulsos nervosos. Suas características básicas são: a) início, geralmente subagudo ou insidioso; b) fraqueza muscular que predomina nos músculos oculares (sem afetar as pupilas), nos músculos faciais, laríngeos e faríngeos e no diafragma;c) fadiga muscular rápida e fácil ao repetir movimentos ou tentar manter a postura, recuperação parcial após um período de repouso e melhora perceptível na resposta à administração de agentes colinérgicos (neostigmina), ed) não há atrofia muscular ou alteração de reflexos, ou outras anormalidades neurológicas, e o principal risco é a facilidade com que o indivíduo desenvolve os episódios; Parada respiratória. Há uma forma idiopática, a mais frequente, e outras associadas a tumores do timo, hipertireoidismo e doenças do colágeno.
Síndrome mieloproliferativa . Nome geral para um grupo de doenças caracterizadas pela proliferação descontrolada de tipos de células na medula óssea. Define os sinais e sintomas de processos evolutivos não agudos que se manifestam pela proliferação predominante, em vez de isolada, de células da linha granulocítica, megacariocítica, eritróide e, eventualmente, de fibroblastos. São eles: a) leucemia granulocítica crônica, que se apresenta com grande esplenomegalia e número de granulócitos circulantes que costuma chegar a 200.000 por mm 3 ; b) trombocitemia hemorrágica essencial, onde os megacariócitos predominam claramente e a esplenomegalia é moderada;c) policitemia vera, caracterizada por um aumento no número de células circulantes das três séries, mas principalmente às custas de eritrócitos e com hematócritos que geralmente excedem 60%, ed) metaplasia mieloide agnogênica ou metaplasia mieloide -mielofibrose, uma condição em que predominam granulócitos e megacariócitos, mas com fibrose progressiva simultânea da medula óssea, de modo que nos estágios avançados quando essa fibrose se completa, a proliferação celular ocorre em focos extramedulares (metaplasia) e explica a notável hipertrofia do fígado e baço que esses pacientes apresentam. Essas quatro pinturas têm em comum,
Síndrome de Milroy-Nonne-Melge . Síndrome do linfedema crônico bilateral dos membros inferiores. É caracterizada por edema crônico e progressivo das pernas, que se inicia nos primeiros estágios da vida, às vezes associado a hemangiomas e linfangiomas. A condição é hereditária, autossômica dominante; predomina em mulheres e se deve à hipoplasia linfática congênita.
Síndrome de Mikulicz . Síndrome de hipertrofia das glândulas salivares e lacrimais. Aumento simétrico e geralmente insidioso e indolor das glândulas salivares e lacrimais, que é acompanhado por uma supressão parcial ou completa concomitante da secreção de lágrimas e saliva. Existem formas idiopáticas e outras que aparecem em pacientes com sarcoidose, linfomas, infecções específicas, etc. A etiologia é desconhecida.
Síndrome de Milkman . Síndrome típica de certas formas de osteoporose grave na idade madura. É observada predominantemente em mulheres e é caracterizada por a) descalcificação difusa com alterações esqueléticas significativas (cifose acentuada, esmagamento vertebral, deformação óssea); b) dores nas costas e nos membros inferiores, muitas vezes do tipo radicular; c) aumento da fraqueza e astenia ed) anormalidades características nas radiografias ósseas (presença de bandas transversais radiotransparentes circundadas por uma borda opaca). A etiologia da doença é desconhecida.
Síndrome de Minkowski-Chauffard . Síndrome de esferocitose hereditária. É caracterizada por anemia leve a moderada, esplenomegalia e icterícia devido ao aumento da bilirrubina não conjugada, devido à presença de esferócitos e aumento da fragilidade dos glóbulos vermelhos. É transmitido como um traço autossômico dominante.
Síndrome de Möbius . Síndrome neurológica de disgenesia dos núcleos dos nervos cranianos. Manifesta-se por tipos e graus de paralisia ou paresia que variam de acordo com o maior ou menor envolvimento dos diferentes nervos cranianos, inexpressividade da face, alterações da motilidade ocular, dificuldade de sugar, disfagia; disartria, etc. Ocasionalmente, esses pacientes apresentam sindactilia e atrofia do grupo muscular. A causa é desconhecida.
Síndrome de Von Monakow . Síndrome neurológica de oclusão da artéria coróide anterior. Hemiplegia alternada acompanhada de hemianopia e hemianestesia homônima ou hemi-hipoestesia devido a lesões que afetam o braço posterior da cápsula interna.
Síndrome de Mondor . Síndrome das formas graves de aborto séptico. É caracterizada por picos febris, icterícia de rápido aumento, hemoglobinúria hipotensiva acentuada e oligúria que quase sempre evolui para insuficiência renal aguda. A fácies geralmente é típica: icterícia conjuntival, bochechas pálidas e cianose perioral.
Síndrome de Morgani-Stewart-Morel . Síndrome de hiperostose frontal interna. Consiste no espessamento da tábua interna do osso frontal que atinge quase que exclusivamente o sexo feminino, próximo à menopausa, e é acompanhado por obesidade, hipertricose e diferentes manifestações neuropsíquicas. É de causa desconhecida.
Síndrome de Morquio , síndrome mucopolissacaridose IV. É caracterizado por: a) fácies com feições moderadamente ásperas, nariz largo; boca larga e alterações dentárias; b) pescoço e altura baixos, cifose acentuada e alargamento ântero-posterior do tórax; c) genu valgo com marcha de pato; d) hepatoesplenomegalia e eventualmente insuficiência aórtica; ee) presença de opacidades corneanas, mas sem retardo mental. É transmitido como um traço autossômico recessivo e é devido a uma deficiência de sulfato de condroitina sulfatase.
Síndrome de Moschcowitz . Síndrome purpúrica com trombose disseminada. O quadro é de uma anemia hemolítica grave do tipo extracorpuscular, associada a púrpura trombocitopênica e distúrbios neurológicos múltiplos (paralisia dos nervos cranianos, sinais cerebelares, vertigens e convulsões). A causa é desconhecida.
Síndrome de mucopolissacaridose . Síndrome de deficiência congênita de enzimas específicas do metabolismo da mucopolissacaridose. Inclui um conjunto de quadros clínicos que têm em comum: a) deficiência congênita de algumas das enzimas essenciais para a degradação de três tipos de mucopolissacarídeos: sulfato de heparan, sulfato de dermatan e sulfato de queratan; b) a natureza progressiva da doença, cujas manifestações são decorrentes do acúmulo crescente de mucopolissacarídeos nos diferentes sistemas corporais, ec) a existência de certas semelhanças básicas quanto ao fenótipo: características grosseiras, disostose, múltipla, rigidez articular, hepatoesplenomegalia, excreção de mucopolissacarídeos na urina, etc.Em vez disso, eles diferem entre si pela gravidade das alterações esqueléticas, pela presença ou ausência de certos componentes particulares (retardo mental, opacidades na córnea), pela gravidade do prognóstico e devido à deficiência enzimática de cada caso . A partir das combinações desses elementos, o grupo geral de mucopolissacaridoses foi subdividido nos seguintes tipos particulares: tipo IH ou síndrome de Hurler; tipo IS ou síndrome de Scheie (anteriormente: tipo V); tipo II ou síndrome de Hunter; tipo III ou síndrome de Sanfilippo; tipo IV ou síndrome de Morquio; tipo VI ou síndrome de Maroteaux-Lamy; tipo VII, ou síndrome de Sly; e tipo VIII ou síndrome de Di Ferrante. ou síndrome de Sanfilippo;tipo IV ou síndrome de Morquio; tipo VI ou síndrome de Maroteaux-Lamy; tipo VII, ou síndrome de Sly; e tipo VIII ou síndrome de Di Ferrante. ou síndrome de Sanfilippo; tipo IV ou síndrome de Morquio; tipo VI ou síndrome de Maroteaux-Lamy; tipo VII, ou síndrome de Sly; e tipo VIII ou síndrome de Di Ferrante.
Síndrome de Morte Súbita Infantil . Nome pelo qual os casos de morte inesperada e inexplicada de uma criança são designados. Normalmente, este é o caso da criança que adormece pacificamente à noite e na manhã seguinte é encontrada morta em seu berço (morte no berço, dos autores saxões), sem qualquer explicação ou achado que o justifique, em estudos de autópsia . A causa é desconhecida, embora tenham sido postulados diversos mecanismos que poderiam ser responsáveis por uma condição desse tipo: a incapacidade da criança, devido à imaturidade, de aumentar a ventilação em resposta a obstruções respiratórias moderadas; intervenção de uma condição como apnéia do sono em adultos; desencadeamento de arritmias cardíacas e outros.
Síndrome de Munchhausen . Síndrome de fabulação em uma personalidade histriônica. É característica de indivíduos que solicitam atendimento em hospitais alegando estar sofrendo de doença aguda, sobre a qual contam uma história coerente, muitas vezes dramática, mas falsa, e que reiteram esse comportamento em diferentes situações de suas vidas.
Síndrome do coto cístico . Síndrome pós-operatória de colecistectomias. Inclui um conjunto de sintomas mais ou menos imprecisos, semelhantes aos da síndrome pós-colecistectomia, exceto pela ausência de icterícia, que estão associados à dilatação do coto residual e são atribuídos a fenômenos reflexos originados nesta área.
Síndrome de Naegeli . Síndrome caracterizada pela associação de hiperceratose, anormalidades pigmentares e outras doenças cutâneas. Inclui hiperceratose palmoplantar, pigmentação reticulada da pele, anidrose ou hipoidrose e, às vezes, alterações dentárias. É hereditário e transmitido como traço autossômico dominante.
Síndrome de Nelson . Síndrome após certos tratamentos para a doença de Cushing. É observada em pacientes com doença de Cushing submetidos a adrenalectomia bilateral com irradiação da hipófise. Consiste no desenvolvimento de um tumor secretor de ACTH mais agressivo, que produz graves alterações do campo visual devido à invasão local.
Síndrome de neoplasias endócrinas múltiplas . Conjunto de manifestações que resultam da hiperfunção simultânea de diversos sistemas endócrinos. Designa uma variedade de quadros clínicos associados à presença de hiperplasias, ou múltiplos tumores endócrinos (pancreáticos; paratireoides, hipofisários, tireoidianos e adrenais, além de neuromas) e cujas manifestações são decorrentes da hiperfunção das glândulas correspondentes. Embora as variedades e combinações teóricas possíveis sejam muitas, as formas específicas de apresentação permitiram estabelecer três grupos distintos dentro da síndrome das neoplasias endócrinas múltiplas: tipo I ou síndrome de Wermer.em que ocorrem tumores de tireoide, adrenal e hipófise, hiperplasia de paratireoide e rumores não insulínicos das ilhotas de Langerhans, e aos quais tende a ser incorporada a síndrome de Zollinger-Ellison, anteriormente considerada uma entidade separada; o tipo II ou síndrome de Sipple, caracterizado pela associação de carcinoma medular de tireoide; com feocromocitomas e hiperplasia da paratireóide; e tipo III ou síndrome de neuroma de mucosa múltipla, uma combinação de um feocromocitoma e um carcinoma medular da tireóide com numerosos neuromas de mucosa e diferentes anormalidades corporais.
Síndrome neuroanêmica . veja síndrome de Lichtheim .
Síndrome de neuroma múltiplo mucoso . Síndrome do tipo III de neoplasias endócrinas múltiplas. É definida pela associação de tumores da tireoide e adrenal (carcinoma medular da tireoide, feocromocitoma), pelo desenvolvimento de múltiplos neuromas na mucosa (boca, conjuntiva, esôfago, trato digestivo) e pela presença de malformações corporais (hábito semelhante a Síndrome de Marfan, cifoescoliose, genu valgo, pés cavos, prognatismo dos tecidos moles, etc.).
Síndrome de neuropatia retrobulbar . Síndrome da cegueira reversível de etiologia desconhecida Manifesta-se em adolescentes ou adultos jovens por uma diminuição repentina da acuidade visual em um olho, que evolui rapidamente para a cegueira, seguida pelo mesmo quadro algumas semanas depois no outro olho. A pupila e a retina estão normais e não há alterações neurológicas que justifiquem o distúrbio. A maioria dos pacientes se recupera espontaneamente, sem sequelas ou com defeitos menores ou pontuais no campo visual.
Síndrome do nevo basocelular . veja síndrome de Gorlin-Goltz .
Síndrome do nó sinusal doente . veja Síndrome da doença do nó sinusal .
 Síndrome de Noonan |
Síndrome de Noonan . Síndrome hereditária caracterizada por hipogonadismo, malformações cardíacas e outras anormalidades corporais. Suas características básicas são: a) altura e pescoço curto, com hipertelorismo, baixa implantação dos cabelos e tórax em escudo; b) hipogonadismo e criptorquidia; c) estenose pulmonar congênita e, ocasionalmente, estenose aórtica, ed) em alguns casos, retardo mental leve ou moderado. Não há alterações no cariótipo.
Síndrome de Nothnagel . Síndrome neurológica do tronco cerebral com distúrbios cerebelares e envolvimento do terceiro nervo craniano. É caracterizada por paralisia parcial unilateral ou bilateral da motilidade ocular (III nervo craniano), associada à ataxia cerebelar (lesão do pedúnculo cerebelar superior). Geralmente, é causada por tumores no teto do mesencéfalo.
Síndrome do núcleo ambíguo e do trato espinotalâmico . veja síndrome de Avellis .
Síndrome do núcleo de Deiters . veja síndrome de Bonnier .
Síndrome de obstrução arterial aguda . Síndrome da interrupção repentina do fluxo arterial para um setor do organismo. É caracterizada por dor, frio, palidez, anestesia, impotência funcional e desaparecimento do pulso periférico. É devido a um espasmo ou à presença de um obstáculo orgânico em uma artéria (trombose, embolia, compressão exógena).
Síndrome da enxaqueca oftalmoplegia . Síndrome oftalmológica transitória associada a crises de enxaqueca. Ocorre em adultos jovens na forma de um ou mais ataques unilaterais de paralisia, de movimentos oculares (nervos cranianos III e VI) associados à enxaqueca, condição que não difere em nada da forma clássica de crise de enxaqueca. Os pacientes curam espontaneamente e a causa é desconhecida, embora a síndrome tenha sido atribuída a espasmo prolongado de algum ramo da artéria oftálmica.
Síndrome de Ogilvie . Síndrome de obstrução aparente do cólon. Manifesta-se por estados de contratura persistente da musculatura do intestino grosso, sem causa orgânica que os justifique. É atribuído a anormalidades da inervação simpática.
Síndrome de Oppenheim . Síndrome das condições que evoluem com flacidez muscular no paciente pediátrico. Nome geral, puramente clínico e independente da etiologia, de todas as afecções do recém-nascido e da infância que se apresentam com diminuição do tônus dos músculos esqueléticos, e das quais a síndrome de Werdnig-Hoffinann é a entidade mais conhecida.
Síndrome de Ostrum-Frost . Síndrome caracterizada por malformações craniocervicais e da cintura escapular . É uma tríade que consiste em sinostose congênita do pescoço, intussuscepção occipital com protrusão das vértebras cervicais na fossa posterior (platibasia) e elevação congênita da escápula (deformidade de Sprengel).
Síndrome do ovário policístico . veja síndrome de Stein-Leventhal
Síndrome de Pancoast . Síndrome neuromuscular de tumores pulmonares de vértice. É definida como um quadro de dor reumática em membro superior, às vezes com atrofia muscular focal, acompanhada de miose e paralisia do elevador da pálpebra superior. É típico de neoplasias próximas ao ápice pulmonar que invadem as raízes do plexo braquial.
Síndrome da pancreatite crônica . Síndrome de inflamação crônica do pâncreas. Manifesta-se por: a) episódios recorrentes de dor abdominal leve a moderadamente intensa de duração variável (horas ou dias); b) esteatorreia e diabetes latente ou manifesto, de instalação insidiosa e evolução progressiva ec) presença de depósitos de cálcio em todo o órgão, visíveis nas radiografias. A condição predomina em alcoólatras, obesos ou indivíduos com litíase biliar, embora também existam formas idiopáticas.
Síndrome de pancreatite hereditária . Síndrome de pancreatite infantil. É definida pelo aparecimento de episódios repetidos de dor abdominal intensa, com duração de dias a semanas, com aumento dos níveis plasmáticos de amilase e lipase, e pelo desenvolvimento progressivo das manifestações crônicas da doença: diabetes, esteatorreia e calcificação do pâncreas . A condição é hereditária e é transmitida como um traço autossômico dominante com penetração incompleta.
Síndrome de Papp-Parkinson . Síndrome de taquicardia paroxística repetitiva. É caracterizada pelo aparecimento de episódios curtos muito numerosos de taquiarritmia atrial paroxística (taquicardia supraventricular, flutter atrial, fibrilação atrial), separados por também intervalos curtos de ritmo sinusal com extrassístoles atriais frequentes. O quadro é de causa desconhecida, pode se repetir até 1000 vezes ao dia e leva o indivíduo a um estado de virtual incapacidade.
Síndrome de paralisia periódica familiar . Síndrome de paralisia intermitente com hipocalemia associada. É caracterizada por ataques recorrentes de paralisia flácida dos músculos dos membros, com arreflexia tendínea e falta de resposta muscular à estimulação elétrica. Os ataques geralmente ocorrem à noite, geralmente após a ingestão de refeições ricas em carboidratos, e são geralmente acompanhados por uma diminuição acentuada nos níveis de potássio plasmático. É uma condição familiar, com mecanismo ainda pouco conhecido, que permite ao indivíduo desenvolver uma vida normal nos períodos intercríticos.
Síndrome paramediana de Foix . veja Síndrome de Dejerine , def. 2
Síndrome paraneoplásica . Conjunto de manifestações associadas à presença de tumor maligno em órgãos e sistemas não afetados diretamente pelo crescimento neoplásico. Inclui diferentes condições que muitas vezes precedem ou acompanham a manifestação clínica do tumor e incluem: a) síndromes endócrinas (Cushing, ginecomastia, hipertireoidismo, hipoglicemia, hiperpigmentação, hiperparatireoidismo, etc.) b) osteoartrofia reumatiforme (polimiosite, poliartrite hipertrófica, etc.); c) neurológicas (doenças cerebelares ou cerebrais), d) hematológicas (trombose disseminada) e outras. Exemplos disso são a associação de hiperparatireoidismo com tumores renais, pancreáticos ou ovarianos; ginecomastia ou hiperpigmentação com carcinomas de pulmão;hipertireoidismo com coriocarcinoma ou com carcinoma embrionário do testículo; e acantose nigricans com linfomas ou adenocarcinomas.
Síndrome paratrigeminal de Raeder . Síndrome de dor unilateral da face. É caracterizada por crises unilaterais de intensa dor facial, localizadas na região maxilar e frontotemporal, acompanhadas de hipoestesia e desigualdade pupilar. Ao contrário da neuralgia do trigêmeo, é causada por lesões orgânicas (tumores; granulomas).
Síndrome de Parinaud , síndrome neurológica, do tronco encefálico com comprometimento dos nervos cranianos. É caracterizada por paralisia do olhar para cima, pupilas fixas e olhos divergentes. É causada por lesão dos tubérculos quadrigêmeos anteriores e do mecanismo de coordenação do olhar supranuclear para cima, e a causa geralmente é um pinealoma.
Síndrome de Parkinson . S índrome de paralisia agitante neurológicas. É um início insidioso e evolução progressiva cujas características básicas são: a) típica fácies seborreica inexpressiva com salivação exagerada; b) rigidez muscular generalizada; c) tremor espesso involuntário, presente em repouso, mas que desaparece durante o sono e tende a desaparecer com os movimentos; d) marcha também característica (marcha festiva), rápida, com passos curtos e com o corpo inclinado para frente. Não há alterações no intelecto ou nos órgãos dos sentidos.É causada por lesão dos núcleos pigmentados do tronco cerebral (substantia nigra, locus coeruleus) e é observada como uma entidade pura na doença de Parkinson e associada a outras manifestações em condições de etiologia diferente (parkinsonismo aterosclerótico, parkinsonismo pós-encefalítico, etc.).
Síndrome de Paterson-Kelly . veja síndrome de Plummer-Vinson .
Síndrome do pedículo de Claude . Síndrome neurológica do tronco encefálico com envolvimento do terceiro nervo craniano e alterações cerebelares e da articulação da palavra. É caracterizada por paralisia da motilidade ocular do lado da lesão (III nervo craniano), acompanhada de hemiasinergia do lado oposto (lesão do núcleo vermelho) e disartria. É causada por aneurismas, trombose ou tumores do teto do cérebro.
Síndrome de Pellegrini-Stieda . Síndrome de disfunção da articulação do joelho. Dor crônica, inchaço e limitação dos movimentos do joelho, com calcificação do ligamento colateral interno dessa articulação. É uma sequela distante de trauma ou hematoma traumático.
Síndrome de Pende . Síndrome de hiperfunção global da adenohipófise. Manifesta-se por: a) aumento moderado da altura; b) tendência à obesidade, preferencialmente na altura dos quadris e coxas; c) hiperplasia mamária em mulheres e ginecomastia em homens; d) sinais de craniopatia metabólica ee) estrias cutâneas: semelhantes às da doença de Cushing. Não há pressão alta e a causa do distúrbio é desconhecida.
Síndrome de Pendred . Síndrome hereditária caracterizada por surdez e alterações na função tireoidiana. É manifestada por surdez congênita bilateral e pelo desenvolvimento posterior na primeira infância de um bócio não hipotireoidiano. O distúrbio é visto em regiões onde não há bócio endêmico e sua causa é desconhecida, embora seja atribuída a uma incapacidade congênita de sintetizar os hormônios da tireoide normalmente.
Síndrome de Penfleld . Síndrome de hiperfunção neurovegetativa episódica associada a disritmia cerebral. É caracterizada por breves acessos de inquietação, com fenômenos vasomotores no território simpático cervical, hipertensão arterial, diaforese, lacrimejamento, miiose pupilar ou midríase, taquicardia, bradipnéia e, às vezes, perda de consciência. Foi atribuída por Penfield a descargas epilépticas originadas no núcleo dorsal do tálamo.
Síndrome perisylviana posterior . veja Síndrome de Afasia Sensorial
Síndrome de Peutz-Jeghers . Síndrome hereditária caracterizada por alterações cutâneas e tendência ao desenvolvimento de processos malignos. Manifesta-se pela presença de máculas pigmentadas nos lábios, na mucosa bucal e nos dedos, e por uma polipose intestinal que predomina no intestino delgado. A condição é transmitida como um traço autossômico dominante e foi demonstrada uma tendência aumentada para o desenvolvimento de adenocarcinomas gástricos, duodenais e de cólon.
Síndrome de Pick . Síndrome de pericardite constritiva crônica. É um quadro de hipertensão intrapericárdica crônica, consecutiva a pericardite reumática ou tuberculosa, na qual há fibrose hepática e ingurgitamento jugular, mas sem batimentos cardíacos ou oscilações respiratórias. A fluoroscopia não mostra os movimentos característicos da atividade cardíaca ("coração parado").
 Síndrome de Pickwick |
Síndrome de Pickwick . Síndrome de hipoventilação pulmonar associada à obesidade. É caracterizada basicamente por obesidade, sonolência diurna acentuada, policitemia e apetite exagerado, com aumento da PCO 2 arterial e sem sinais de doença pulmonar orgânica de base; além disso, geralmente há cianose e respiração noturna periódica e, eventualmente, sinais de insuficiência cardíaca direita. As manifestações melhoram acentuadamente, quando o paciente reduz o peso.
Síndrome das pernas inquietas . Síndrome de uma das variedades de insônia. É caracterizada por: a) a necessidade virtualmente irreprimível do indivíduo de movimentar as pernas quando imóvel ou sentado, e muito particularmente quando deitado e tenta dormir; b) a explicação que fornece sobre esses movimentos, que relaciona com um desconforto profundo, intenso e extremamente desagradável que sente nos tornozelos, e do qual tenta se libertar movendo as pernas; c) o desaparecimento da sensação após o sono; ed) em todos os casos, a possibilidade de registrar, durante o sono, movimentos rítmicos estereotipados, dorsiflexão dos pés e dedos dos pés (movimentos periódicos do sono), característicos de diferentes tipos de insônia .A causa é desconhecida, embora seja atribuída a alterações neurofisiológicas reais, embora ainda não completamente conhecida.
Síndrome pineal . Síndrome em que aparecem certos tumores do timo. É caracterizada por crescimento anormal precoce de ossos longos, desenvolvimento precoce da genitália externa e função sexual e hipertensão intracraniana.
Síndrome do assoalho orbital. veja síndrome de Dejean .
Síndrome de Plummer-Vinson . Síndrome de anemia hipocrômica idiopática com acílios. É observada quase exclusivamente em mulheres e se manifesta por: a) anemia hipocrômica que responde satisfatoriamente à administração de altas quantidades de ferro; b) acílios gástricos; c) atrofia da mucosa da boca, faringe e esôfago superior, com disfagia e glossite; e caracteristicamente, d) presença de prega em forma de lua crescente na região cricofaríngea. A etiologia da doença é desconhecida.
Síndrome pós-colecistectomia . Síndrome vagamente definida em alguns pacientes submetidos à colecistectomia. Conjunto de sintomas que costumam ser vagos e mal definidos, incluindo dor difusa ou com cólica ou desconforto no quadrante superior direito do abdome, intolerâncias alimentares, sensação de saciedade etc. e, ocasionalmente, icterícia. Em sentido estrito, o nome deve ser aplicado apenas nos casos em que se demonstre que as manifestações são decorrentes de operações incompletas (cálculos residuais), de omissões intraoperatórias (neoplasias que passaram despercebidas) ou de complicações específicas (estenose pós-operatória) .No entanto, por costume e não por extensão, também se aplica a pacientes com queixas que sugerem um histórico emocional ou funcional apenas porque foram submetidos à cirurgia, e a outros que se queixam dos mesmos sintomas que tinham antes da cirurgia. intervenção,
Síndrome pós-comissurotomia . Síndrome pós-operatória longe de comissurotomia por estenose mitral. É um quadro relativamente frequente neste tipo de operação, que aparece um a três meses após a intervenção e se manifesta por febre, dor retroesternal, artralgias difusas, muitas vezes migratórias, arritmias cardíacas; sinais de polisserosite (pericardite, pleurisia), etc. A causa não foi precisamente estabelecida.
Síndrome pós-flebítica . veja Síndrome pós-trombótica .
Síndrome pós-gastrectomia . veja síndrome de esvaziamento rápido
Síndrome pós-infarto do miocárdio . veja síndrome de Dressler .
Síndrome pós-radiação . veja Síndrome de radiação .
Síndrome pós-perfusão . Síndrome de infecção por citomegalovírus. É um quadro febril agudo com hepatomegalia, esplenomegalia e presença de linfócitos atípicos, inicialmente observado em pacientes submetidos a intervenções cardíacas com circulação extracorpórea. Posteriormente, foi demonstrado que o agente causador era um citomegalovírus e que as possíveis vias de transmissão eram muitas.
Síndrome pós-quicárdica . Síndrome eletrocardiográfica residual de taquicardias ventriculares. É observada após crises prolongadas e é caracterizada pelo aparecimento de ondas T ampliadas e invertidas que persistem por um tempo variável (horas ou dias) antes de finalmente desaparecerem. É uma manifestação do processo de recuperação funcional e carece, por si só, de significado patológico.
Síndrome pós-trombótica . Síndrome crônica de trombose venosa de membros inferiores. É uma condição dermatológica observada em pacientes que sofreram trombose de repetição, caracterizada por uma primeira fase em que predominam edema e hemorragias subcutâneas; uma segunda fase com hiperpigmentação de hemossiderina, fibrose subcutânea, atrofia da pele e obstrução linfática; e uma terceira fase em que aparecem as úlceras: crônicas, recorrentes e de difícil cicatrização.
Síndrome de Pourfour du Petit. Síndrome de irritação cervical simpática. É o oposto da síndrome de Horner e se manifesta por exoftalmia, midríase e aumento da fenda palpebral. É o resultado de um fenômeno irritativo, geralmente devido à compressão ou infiltração tumoral.
Síndrome do pré-motor . Síndrome neurológica das lesões do córtex cerebral pré-motor. Manifesta-se como hemiplegia espástica com hiperreflexia osteotendínea, perda ou diminuição da capacidade de realizar movimentos finos, distúrbios vasomotores transitórios e atitudes do tipo apreensão compulsiva. É produzida, quase sempre, por processos tumorais expansivos.
Síndrome de Profichet . Síndrome caracterizada por calcificações subcutâneas e alterações musculares. Manifesta-se pela formação gradual de nódulos de cálcio sob a pele, especialmente ao redor das grandes articulações, seguido por atrofia muscular, ulcerações cutâneas e distúrbios neurológicos. Predominam em jovens e a causa não foi estabelecida.
Síndrome pontina superior . veja síndrome de Raymond-Cestan .
Síndrome de Putnam-Dana . veja síndrome de Lichtheim .
Síndrome de QT. Síndrome caracterizada por anormalidades eletrocardiográficas e episódios de taquiarritmia paroxística. Aplica-se aos casos em que um intervalo QT anormalmente prolongado, de natureza congênita e presente em ritmo sinusal normal, está associado a episódios paroxísticos de taquicardia ventricular polimórfica ("torsade de points"), em que o trânsito do ritmo sinusal para a arritmia é caracterizado pela deformação cada vez mais acentuada dos sucessivos complexos QRS, até que o padrão característico de taquicardia ventricular seja estabelecido. Inclui as síndromes de Jervell e Lange-Nielsen, de transmissão autossômica recessiva, acompanhada de surdez nervosa, e a de Romano-Ward, de herança autossômica dominante e sem surdez nervosa.
Síndrome quiasmática . Síndrome das lesões do quiasma óptico. Manifesta-se por cefaleia, tonturas e desmaios, perda progressiva da visão, muitas vezes não sistematizada e dependendo da maior ou menor extensão em que os diferentes feixes são afetados; e redução do campo visual. A causa é o tumor.
Síndrome de Ramsay-Hunt . Síndrome de afecções viróticas do gânglio geniculado. Manifesta-se por severa paralisia facial unilateral, acompanhada de dor de ouvido e erupção vesicular, em feixe, acometendo conduto auditivo externo e pavilhão auricular. É devido à lesão do gânglio geniculado causada pelo vírus herpes zoster.
Síndrome de Raymond-Cestan . Síndrome neurológica de lesão de Pons. É caracterizada, no lado oposto da lesão, por hemiparesia, hemianestesia, tremor estático e assinergia do tipo cerebelar, e pela paralisia dos movimentos de lateralização do olhar que predomina no lado da lesão. Geralmente é de origem vascular e é causada por lesões que afetam a parte superior da ponte anular.
Síndrome de Raynaud . Síndrome caracterizada por surtos de vasoconstrição paroxística. Designa um quadro de episódios isquêmicos paroxísticos, principalmente dos dedos das mãos, que se apresentam caracteristicamente como uma reação trifásica: primeiro palidez, depois cianose e, por fim, vermelhidão, muitas vezes com dor e parestesia na primeira fase. Do ponto de vista clínico e para a mesma síndrome, é necessário separar a doença de Raynaud, um distúrbio funcional benigno e simétrico que predomina em mulheres jovens e geralmente é desencadeado por frio ou emoção, do fenômeno de Raynaud secundário, que acompanha outras doenças patológicas processos.Este último não tem distinção de sexo e idade, pode ser assimétrico, às vezes pode causar úlceras digitais, e é observado em certas colagenopatias; (esclerodermia), no envenenamento por chumbo e arsênio, durante a administração de diferentes medicamentos (ergotamina metisergida, propranolol), nas doenças hematológicas; (por aglutininas frias); em carcinomas ocultos, etc.
Síndrome de Reichmann . Síndrome de hipersecreção gástrica. Designa o estado caracterizado por hipersecreção de suco gástrico ácido, qualquer que seja sua causa e independentemente de eventuais complicações.
Síndrome de Reifenstein . Síndrome de pseudo-hermafroditismo masculino associada a hipogonadismo primário. É caracterizada por eunucoidismo, ginecomastia e hipospádia e por atrofia dos túbulos seminíferos com ausência de esperma. O distúrbio é devido a anormalidades na síntese ou na ação periférica da testosterona, se manifesta na idade pós-puberal e é transmitido como um traço recessivo ligado ao X.
Síndrome de Reye . Síndrome paravirótica infantil com distúrbios neurológicos e renais hepáticos. Ela ocorre em crianças previamente saudáveis com infecções do trato respiratório superior e é caracterizada por: a) sinais de dano hepatocelular grave, com hipoglicemia, acidose, hiperamonemia e aumento acentuado das transaminases séricas; b) encefalopatia grave com vômitos, edema cerebral e alterações rapidamente progressivas do sistema nervoso central; c) presença de extrema vacuolização de células hepáticas e túbulos renais ed) alta mortalidade, mas com recuperação complexa nos pacientes que sobrevivem.É observada como uma complicação de infecções virais (devido a vírus; influenza A, varicela, grupo Coxsackie e outros) cujo curso inicial não difere do usual para cada tipo de doença, e quase sempre em pacientes que foram tratados com aspirina. Por esta razão,
Síndrome de Riley-Day . Síndrome de disautonomia familiar. É caracterizada por instabilidade autonômica (transpiração anormal, perda do controle vasomotor, hipertensão lábil), distúrbios do paladar, diminuição das sensações de dor e temperatura, hiporrefexia, febre episódica, convulsões, vômitos e falta de lacrimação. A maioria das pessoas afetadas não chega à idade adulta. Seria devido a uma deficiência enzimática ainda não especificada.
Síndrome do roubo da subclávia . Síndrome de isquemia no território vertebrobasilar, decorrente de estenose - antes do nascimento da vértebra - de uma das artérias subclávia. É caracterizada pelo aparecimento de tontura, síncope ou outras manifestações de insuficiência vertebrobasilar, quando o paciente faz exercícios com o braço do lado afetado. Nessas circunstâncias, em que a subclávia não consegue fornecer o sangue que o membro ativo necessita, ocorre uma inversão de fluxo compensatório no sistema vertebral (do cérebro passa para a subclávia, além da oclusão, e daí o braço) e sintomas aparecem; isto é, a subclávia "rouba" o sangue que deveria ir para o cérebro.
Síndrome de Rochon-Duvigneau . Síndrome neurológica de lesão unilateral dos nervos cranianos III, IV, V e VI. Geralmente é uma condição insidiosa e progressiva que leva à oftalmoplegia completa (nervos cranianos III, IV e VI), com anestesia da córnea (ramo oftálmico do nervo craniano V). É causada por aneurismas, meningiomas e outros tumores que crescem nas proximidades da fenda esfenoidal
Síndrome de Roger . Síndrome de tumores malignos do esôfago. Designa o quadro de salivação contínua que acompanha as neoplasias do esôfago ou da região esofagofaríngea.
Síndrome de Romano-Ward . veja Síndrome QT
Síndrome de Rothmann-Makai . Síndrome de inflamação circunscrita do tecido adiposo subcutâneo. Designa um quadro benigno de paniculite circunscrita, manifestada por necrose de células de gordura e formação de granulomas adiposos que se transformam em estruturas císticas. A causa é desconhecida.
Síndrome de Rothmund-Thomson . Síndrome esclerodérmica familiar com telangiectasias cutâneas e múltiplas alterações orgânicas. É caracterizada pelo desenvolvimento de: a) esclerose em faixas simétrica e progressiva que predomina na face e nos dedos e rapidamente leva à rigidez; b) telangiectasias cutâneas com alterações pigmentares associadas; c) catarata e sinais generalizados de envelhecimento precoce, d) ulcerações tróficas ee) hipogonadismo e outras anormalidades. É transmitido como traço autossômico recessivo.
Síndrome da ruptura iminente do útero . veja síndrome de Bandl-Frommel
Síndrome de Roussy-Lévy . Síndrome neuromuscular com manifestações centrais e periféricas. Designa a combinação de uma atrofia muscular progressiva, de origem neuropática e que predomina na região fibular, com ataxia cerebelar e escoliose da coluna dorsal e lombar.
Síndrome de Sanfilippo . Síndrome de mucopolissacaridose III. É caracterizada, dentro do grupo de: mucopolissacaridose, pelo notável predomínio de alterações neurológicas em relação às somáticas, que são relativamente leves. Suas características básicas são: a) fácies de feições grosseiras, sobrancelhas desgrenhadas, boca entreaberta e múltiplas anomalias dentárias; b) rigidez articular moderada com mínimas alterações esqueléticas (pescoço e dedos curtos); c) hepatoesplenomegalia ed) surdez, retardo mental grave e ausência de opacidades corneanas. É transmitido como traço autossômico dominante, devido à deficiência de diferentes;enzimas (heparansulfatase, N-acetil-D-glucosaminidase, etc.) e quatro variantes diferentes foram descritas: (tipos A, B, C e D).
Síndrome de Sayre-Kearns . Síndrome caracterizada pela associação de miopatia ocular, miopatia miocárdica e retinite pigmentosa. É definida pelo desenvolvimento de oftalmoplegia bilateral progressiva que respeita a pupila e os músculos da acomodação, acompanhada por retinite pigmentosa e miopatia cardíaca concomitante, e pela presença de um número impressionante de mitocôndrias nos tecidos afetados. A causa é desconhecida.
Síndrome de Scheie . Síndrome de mucopolissacaridose IS. É caracterizada por: a) traços faciais ásperos, com boca grande, narinas largas e sobrancelhas peludas; b) rigidez articular que surge por volta dos 10 anos de idade, com distúrbios esqueléticos menores e insuficiência aórtica em fases posteriores da vida; c) presença de opacidades corneanas ed) inteligência normal. É transmitida como um traço autossômico recessivo e se deve, como a síndrome de Hurler, a uma deficiência de alfa-L-iduronidase, mas, ao contrário disso, os indivíduos afetados podem ter uma longa sobrevida.
Síndrome de Schmidt . Síndrome neurológica de lesão unilateral do núcleo ambíguo e acessório do X nervo craniano. É definida pela presença de paralisia das pregas vocais, palato mole e dos músculos trapézio e esternocleidomastóideo, do mesmo lado da lesão. A causa geralmente é vascular ou tumoral
Síndrome de Schwachmann . Síndrome de hipoplasia congênita do pâncreas com alterações hematológicas. A manifestação dominante é um quadro de diarreia com esteatorreia, semelhante ao observado na fibrose cística do pâncreas, acompanhada por alterações hematológicas cuja causa está na medula óssea (neutropenia e, menos frequentemente, anemia e trombocitopenia), e ofuscada por infecções breakthrough que costumam ser graves. Ao contrário da fibrose cística do pâncreas, a concentração de solutos no suor é normal e não há distúrbios respiratórios. É transmitido como um traço autossômico dominante.
Síndrome de Seabright-Bantam . Síndrome de pseudo-hipoparatireoidismo. Suas características básicas são: a) nanismo, obesidade e retardo mental de grau variável; b) fácies arredondada e pescoço, tronco, membros e dedos curtos; c) presença de calcificações disseminadas; d) hipocalcemia, crise de tétano e convulsões; ee) hiperplasia da paratireóide com níveis elevados de hormônio da paratireóide circulante. O quadro é familiar de causa desconhecida e deve-se a uma resistência primária dos órgãos efetores (ossos e rins) à ação do hormônio paratireóideo endógeno.
Síndrome de Selye . veja Síndrome de ajuste geral .
Síndrome de Senear-Usher . Síndrome do pênfigo eritematoso. Consiste em lesões crônicas de aspecto seborréico, que predominam na face e no tronco e geralmente não afetam as mucosas. A evolução é mais longa do que a do pênfigo vulgar.
Síndrome do seio carotídeo . Síndrome de estimulação do seio carotídeo. É um quadro súbito de vertigem, lipotimia ou síncope, às vezes com convulsões, desencadeada por estimulação mecânica acidental de um seio carotídeo (compressão digital, rotação abrupta e exagerada do pescoço). A hipotensão está associada à bradicardia, característica de episódios vasovagais.
Síndrome do seio cavernoso . veja síndrome de Foix .
Síndrome pseudobulbar . Síndrome neurológica associada a lesões difusas do sistema nervoso central . É definida pela presença de múltiplos distúrbios neurológicos e psíquicos, e em particular de mimetismo, deglutição e fonação, produzidos por lesão dos núcleos da base, das regiões faciais do córtex cerebral ou dos feixes geniculados. Em geral, não se apresenta como síndrome pura, mas como parte de um quadro de deterioração cerebral global, produzida por lesões vasculares difusas ou por múltiplos focos de amolecimento.
Síndrome de pseudoclaudicação intermitente . Causa neurológica da síndrome de claudicação da marcha. É um quadro facilmente confundido com o da claudicação intermitente de origem vascular (dor que surge ao caminhar e é aliviada com o repouso), mas da qual difere devido: a) à relação entre a distância percorrida e o aparecimento da dor , que não é constante como no caso da claudicação isquêmica, mas muito variável, e entre o início do repouso e o desaparecimento da dor que não ocorre de forma rápida, mas lenta e em tempo variável; b) a distribuição da dor, que predomina na coxa e região glútea e não nos tornozelos;c) a coexistência de lombalgia mais ou menos permanente, que muitas vezes persiste em decúbito, ed) a normalidade dos pulsos periféricos. A imagem é devida a uma compressão geralmente subaguda e lentamente progressiva das raízes nervosas,
Síndrome de pseudocoma . Síndrome neurológica caracterizada por imobilidade e ausência de resposta a estímulos semelhantes aos do coma, mas com conservação da consciência e produzida pela interrupção praticamente completa das vias eferentes de expressão corporal. Suas características básicas são: a) é uma consequência direta de um acidente vascular cerebral; b) nem espontaneamente nem em resposta a estímulos, há movimentos dos membros do tronco, da face ou da faringe, nem são emitidos sons, como ocorre no coma, mas com a particularidade de persistirem os movimentos de piscar, ec) o indivíduo está consciente, conforme evidenciado pelos movimentos de piscar do examinador em resposta direta às perguntas.A síndrome é produzida pela trombose da artéria basilar na região ventral da ponte, que interrompe todas as vias descendentes do corticosterol e corticobulbar, mas sem afetar o sistema de ativação reticular (consciência) ou as vias de motilidade vertical do olho. (piscar).
Síndrome de pseudo-hipoparatireoidismo . veja Síndrome Seabright-Bantam
Síndrome pseudomiastênica . veja síndrome de Eaton-Lambert
Síndrome de pseudotumor cerebral . Síndrome caracterizada por manifestações neurológicas comparáveis às de um tumor cerebral, mas sem patologia orgânica detectável. Predominam em mulheres jovens e suas características são as seguintes: a) cefaléia moderada, lentamente progressiva, ocasionalmente acompanhada de tontura ou diplopia leve, em pacientes com excelente estado geral; b) papiledema bilateral associado a estreitamento do campo visual e aumento do ponto cego, em ambos os casos de grau leve; c) hipertensão do líquido cefalorraquidiano ed) tamanho normal ou discretamente diminuído dos ventrículos cerebrais, normalidade dos estudos tomográficos e arteriográficos e ausência de elementos atípicos na análise do líquido cefalorraquidiano.A condição é reversível, responde ao tratamento com corticosteroides e sua causa é desconhecida.
Síndrome de pseudoxantoma elástico . veja síndrome de Groenblad-Strandberg .
Síndrome de Sézary . Síndromo da fase leucêmica da micose fungóide. É observada nos estágios avançados da micose fungóide (linfoma cutâneo maligno), quando as células neoplásicas já se encontram no sangue circulante, e é definida pela presença de hiperpigmentação, placas dismórficas e espessamento difuso da pele e, fundamentalmente, de uma eritrodermia generalizada. Nas fases terminais ocorre hepatoesplenomegalia junto com outras manifestações de invasão visceral.
Síndrome de Shy-Dragger . Síndrome de hipotensão ortostática com outras manifestações neurológicas . O quadro é de síndrome de hipotensão ortostática idiopática, à qual se somam as manifestações de lesões cerebrais difusas. É observada quando predomina a degeneração das vias extrapiramidais, dos gânglios da base e do núcleo dorsal do vago.
Síndrome de Sickert-Millikan . Síndrome de oclusão intermitente da artéria basilar. É manifestada por episódios recorrentes, transitórios e mais ou menos fugazes de vertigem, perda de visão, diplopia, disartria, disfagia e, às vezes, hemiparesia.
Síndrome de Silvestrini-Corda . Síndrome hiperestrogênica de doença hepática crônica avançada. Designa o conjunto de sinais de feminização que são observados na cirrose, fígado avançado e que resultam da incapacidade do fígado de inativar os estrogênios circulantes: ginecomastia, hábito eunucóide, perda de pelos pubianos e axilares, diminuição da libido, esterilidade e atrofia testicular.
Síndrome silviana frontal superior . veja Síndrome de Afasia Motora
 Síndrome de Stein-Leventhal. Notar a hipoplasia mamária, tendência à obesidade e virilização em paciente de 16 anos (Surós, Semiología Médica, 1968) |
Síndrome de Sipple . Síndrome do tipo II de neoplasias endócrinas múltiplas. Designa a presença de um feocromocitoma (que geralmente é bilateral e múltiplo), um carcinoma medular da tireoide e uma hiperplasia da paratireoide. Também pode haver outros tumores: meningiomas, glioblastomas, etc. Os sintomas dominantes são geralmente os de hiperparatireoidismo e, menos freqüentemente, os de feocromocitoma. O distúrbio é herdado e transmitido como traço autossômico dominante.
Síndrome de siringomielia . Síndrome neurológica de gliomatose da substância cinzenta da medula espinhal. A siringomielia é uma proliferação da glia periependimal (gliomatose) que destrói a substância cinzenta medular e afeta principalmente os segmentos cervical e lombar. É caracterizada, basicamente, por distúrbios sensoriais (dissociação siringomielica, perda da sensibilidade térmica e dolorosa, com preservação da sensibilidade tátil e profunda), manifestações motoras (paralisia espástica) e alterações tróficas (artropatia siringomielica, atrofias, do tipo Aran-Duchenne , etc.) A causa é desconhecida.
Síndrome de Sjögren . Síndrome caracterizada pela tríade de artrite, xeroftalmia e xerostomia. É uma imagem de olhos secos (xeroftalmia, ceratoconjuntivite seca) e boca seca (xerostomia) que ocorre em indivíduos com artrite reumatóide. Tem curso crônico e é atribuído a mecanismos autoimunes.
Síndrome de Sluder . Síndrome de neuralgia do gânglio esfenopalatino. É caracterizada por dor unilateral maçante e contínua na região da mandíbula superior, que se espalha para o interior do pescoço e ombro, e é acompanhada por obstrução nasal, rinorreia e dor de ouvido.
Síndrome manhosa . Síndrome de mucopolissacaridose VII. É uma forma semelhante à síndrome de Hurler leve e é caracterizada por: a) baixa estatura com peito em quilha; b) dismorfia facial com fácies achatada de feições ásperas, nariz deprimido e sulco subnasal alto; c) retardo mental moderado ed) inclusões marcantes em granulócitos. É causada por uma deficiência de beta-glucuronidase e é transmitida como um traço autossômico recessivo.
Síndrome de Spiller . Síndrome neurológica da compressão crônica da medula espinhal. É uma sequela de paquimeningite incompletamente resolvida (cicatrizes remanescentes) e se manifesta por dor e outros distúrbios sensoriais, fraqueza muscular, paralisia ascendente e sinais de mielite transversa.
Síndrome de Steele-Rlchardson-Olszewski . Síndrome neurológica de paralisia supranuclear progressiva. O início é insidioso e se caracteriza por alterações de equilíbrio, marcha e quedas inesperadas, rigidez afetando principalmente o pescoço e em menor extensão os músculos do tronco, dificuldade de direcionar o olhar para baixo e enfraquecimento do volume da voz. . No estágio final há extrema rigidez, oftalmoplegia completa, anartria e incapacidade total.
Síndrome de Stein-Leventhal . Síndrome do ovário policístico. É observada em mulheres jovens e é definida pela associação de ovários policísticos, oligomenorréia e esterilidade, que são acompanhados por vários graus de obesidade, hirsutismo e hipoplasia mamária. É atribuído a alterações na síntese de estrona.
Síndrome de Stevens-Johnson . Síndrome de uma forma grave de eritema multiforme. É caracterizada pelo desenvolvimento de lesões ulcerativas localizadas na mucosa oronasal e anogenital, olhos e vísceras, com cefaleia, artralgia, febre e prostração. Pode ser fatal. Existem casos idiopáticos e outros associados a doenças virais ou ingestão de diferentes medicamentos.
Síndrome de Still . Síndrome de uma das formas de artrite reumatóide. Refere-se a um quadro de poliartrite crônica acompanhada por adenopatias generalizadas, episódios febris recorrentes com erupção cutânea e esplenomegalia; pode haver pericardite, pleurisia e pneumonite. Há anemia, leucocitose e testes negativos para fator reumatoide e anticorpos antinucleares. As manifestações começam na infância ou adolescência e a causa não é conhecida com precisão.
Síndrome de Stokes-Adams . Síndrome associada ao bloqueio atrioventricular. É caracterizada por perda súbita de consciência, com ou sem convulsões, devido à falha do marcapasso inferior em bloqueio AV completo.
Síndrome de Strachan . Síndrome caracterizada por distúrbios neurológicos e alterações cutaneomucosas de origem nutricional. Define um quadro de desenvolvimento progressivo de doenças oculares (diminuição da capacidade visual, cegueira), sinais de neuropatia periférica (parestesia, ataxia, perda de sensibilidade superficial e profunda) e lesões cutâneas e mucosas (glossite, estomatite, etc.). observada em populações humanas desnutridas ou submetidas a condições forçadas de restrição alimentar (por exemplo, em cidades sitiadas).
Síndrome de Sturge-Weber-Dimitri . Síndrome da angiomatose encefalotrigeminal. É definida como a associação de um angioma facial plano, do tipo capilar e localizado no território de distribuição trigeminal, com um angioma meníngeo venoso, que se localiza mais freqüentemente na região parieto-occipital. Clinicamente, manifesta-se por crises epilépticas extremamente frequentes, às vezes com hemiparesia e hemianopia, e por retardo mental, enquanto as radiografias de crânio mostram a presença de calcificações lineares características.
Síndrome de Sudeck . Síndrome de osteoporose pós-traumática circunscrita. Designa os focos de osteoporose localizada, acompanhada por dor e atrofia dos tecidos moles, que às vezes se desenvolve como consequência de pequenos traumas repetidos (por exemplo, nos dedos dos pés), e mais frequentemente ao redor das áreas de fratura durante o período de imobilização.
Síndrome de Sweet . Síndrome da dermatose neutrofílica febril aguda. É caracterizada por uma erupção não pruriginosa de placas ou nódulos vermelhos, dolorosos e parcialmente confluentes, que começa nos braços e geralmente afeta mulheres; há febre alta e neutrofilia. A causa é desconhecida.
Síndrome tabética . Síndrome neurológica de sífilis avançada. Tabes dorsalis é uma meningoradiculite posterior que causa esclerose rápida das cordas posteriores da medula espinhal. Suas manifestações básicas são: a) ataxia com marcha de calcanhar e hipotonia muscular; b) dissociação da sensibilidade do tipo tabético (perda da sensibilidade profunda e tátil, com preservação da sensibilidade à dor e ao calor); c) alterações tróficas (má perfuração plantar, artropatias tabéticas) ed) sorologia positiva para sífilis.
Síndrome de Takayasu . Síndrome de oclusão progressiva dos ramos do arco aórtico. É caracterizada por uma perda de pulso em ambos os braços e nas carótidas, juntamente com sintomas causados por isquemia cerebral (síncope, hemiplegia transitória, etc.), ocular (cegueira transitória, atrofia retiniana, etc.), da face (músculo atrofia) e braços (claudicação). É decorrente da inflamação e obliteração progressiva do tronco braquiocefálico e das artérias subclávia esquerda e carótida primitiva, acima de sua origem no arco aórtico. Uma etiologia auto-imune é assumida.
 Síndrome de Still |
Síndrome de Tapia . Síndrome neurológica de lesão de dois núcleos de nervos cranianos. Manifesta-se como paralisia unilateral da laringe (núcleo ambíguo) e da língua (núcleo do hipoglosso), sem paralisia do palato mole.
Síndrome de taquicardia paroxística repetitiva . veja síndrome de Papp-Parkinson .
Síndrome de TAR . veja síndrome trombocitopênica com ausência de rádio
Síndrome talâmica . Síndrome de lesão óptica do tálamo. Dependendo de quais regiões são predominantemente afetadas, a síndrome inclui combinações variáveis de algumas das seguintes manifestações que são observadas no lado do corpo, oposto ao da lesão: a) hemianestesia profunda total com hemianestesia superficial menos acentuada; b) dor desagradável e intensa (dor talâmica) no lado afetado, que surge após o período agudo e nos casos em que o núcleo posteroinferior foi acometido; c) alterações motoras, incluindo hemiparesia transitória, redução da motilidade voluntária e aparecimento de movimentos anormais (tremor e ataxia, sincinesias de imitação de movimentos coreicos e atetósicos);d) hemianopia homônima, em lesões posteriores; ee) manifestações neuropsíquicas, com acentuada redução da linguagem, perseverança verbal e ideias, e assim por diante.
Síndrome de Taussig-Bing . Síndrome de doença cardíaca congênita com malformações complexas. É definida por: a) uma tríade anatômica composta pela transposição dos grandes vasos, comunicação interventricular e impulso da artéria pulmonar, que produzem hipertrofia secundária do ventrículo direito; b) uma saturação de oxigênio, que é maior no sangue da artéria pulmonar do que no sangue da aorta ec) suas manifestações clínicas: hipertensão pulmonar, cianose permanente, retardo de crescimento, dedos em coxa, etc.
Síndrome de Thieberge-Weissenbach . Síndrome de esclerodermia com calcificações difusas. É definida pela associação de esclerodermia generalizada com calcinose difusa, que é atribuída à disfunção da paratireoide concomitante.
Síndrome de Thomson . veja síndrome de Rothmund-Thomson .
Síndrome da tíbia anterior . Síndrome de necrose do músculo tibial. Ela se manifesta como um rápido inchaço, aumento da tensão, dor e necrose isquêmica dos músculos tibiais anteriores. A pele torna-se ingurgitada e brilhante, eritematosa e brilhante à medida que a necrose progride. A causa é desconhecida, embora deva estar relacionada ao exercício excessivo.
Síndrome de Tietze . Síndrome dolorosa localizada nas articulações condrocostais. É evidente que predomina em mulheres entre 35 e 50 anos e é caracterizada por dor localizada em uma das articulações condroesternais e, menos freqüentemente, em uma articulação esternoclavicular
Síndrome de Tolosa-Hunt . Síndrome de dor unilateral da face. É caracterizada por dor facial unilateral, claramente predominante na região orbital (primeiro ramo trigeminal), acompanhada de oftalmoplegia e pupilas desiguais. É causada por processos inflamatórios ou tumorais localizados nas proximidades da fenda orbitária superior ou do seio cavernoso.
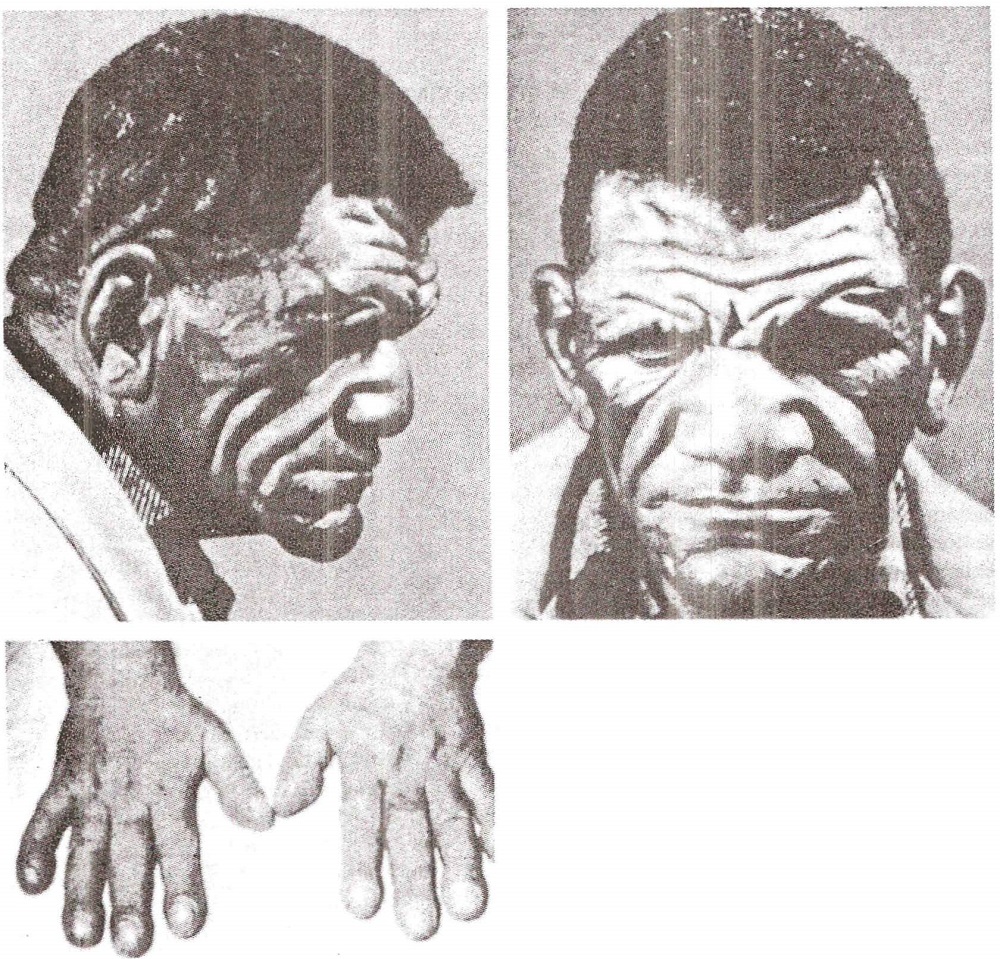 Síndrome de Touraine-Solente-Golé. Observe o acentuado espessamento da pele e hiperplasia de tecidos moles dos dedos com unhas curvas (Modero Medicine, 1973) |
Síndrome de Toni-Fanconi . veja síndrome de Fanconi .
Síndrome de Touraine-Solente-Golé . Síndrome da paquidermoperiostose. Consiste em um espessamento acentuado da pele da face, testa e couro cabeludo, com formação de dobras espessas, neoformação perióstea e desenvolvimento de dedos em coxa. É herdado como um traço autossômico dominante e geralmente aparece na puberdade.
Síndrome de Treacher-Collins . Síndrome de quadro hereditário com alterações predominantemente maxilofaciais. Suas características básicas são: a) hipoplasia bilateral da mandíbula e ossos malar; b) malformações palpebrais, que incluem a presença de coloboma no terço externo da pálpebra inferior ec) anomalias da orelha, às vezes do ouvido médio e interno, e eventualmente surdez. É um distúrbio familiar no qual ocorrem malformações durante a vida intrauterina.
Síndrome tricorrinofalângica . Síndrome caracterizada por anormalidades no esqueleto, cabelo e nariz . É caracterizada por braquidactilia e desvio axial dos dedos, escoliose ou lordose, baixa estatura, cabelos ralos e finos e nariz piriforme com longo sulco subnasal. A causa é desconhecida e compatível com uma vida normal.
Síndrome do X triplo . Síndrome hereditária de indivíduos com retardo mental e amenorréia. Ela ocorre em mulheres e é definida como a associação de fraqueza mental de grau variável, amenorréia secundária e anormalidades corporais incomuns. As pessoas afetadas têm uma trissomia do cromossomo sexual X.
Síndrome de trissomia 21 . veja Síndrome de Down .
Síndrome trombocitopênica com ausência de rádio . Síndrome de quadro combinado de malformações cardíacas e ósseas e alterações hematológicas. É definida pela presença de tetralogia de Fallot ou defeitos no septo interatrial, acompanhada de trombocitopenia e aplasia radial. É uma condição hereditária.
Síndrome de trombose capsular . Síndrome neurológica devido a lesão unilateral da cápsula interna. É a síndrome clássica, completa com hemiplegia alternada, em que ocorre paralisia da metade do corpo e da face do lado oposto à lesão cerebral. É produzido pela oclusão dos ramos perfurantes da artéria cerebral média.
Síndrome de Trousseau . Síndrome trombótica associada à presença de processo neoplásico. Aparecimento de trombose venosa espontânea, geralmente repetida, sem causa aparente e predominantemente em membros inferiores associada à presença de carcinoma oculto. É considerada uma síndrome paraneoplásica.
Síndrome do tubérculo quadrigêmeo . Síndrome neurológica da lesão unilateral dos tubérculos quadrigêmeos. Suas manifestações são basicamente hipoacusia auditiva e ocular do tipo central acompanhada de paralisia dos músculos dependentes do terceiro nervo craniano, paralisia do olhar vertical para cima, paresia de convergência, nistagmo e anisocoria com o sinal de Argyll-Robenson. A causa geralmente é tumor.
Síndrome do túnel do carpo . Síndrome neurológica periférica devido à compressão do nervo mediano. É caracterizada por dor e formigamento ou parestesias em queimação na mão e nos dedos, que podem causar degeneração das fibras nervosas e levar à atrofia muscular. Resulta da compressão do nervo mediano no canal do carpo e costuma ser bilateral.
Síndrome do túnel do tarso . Síndrome neurológica periférica devido à compressão do nervo tibial posterior. É devido à compressão desse nervo no canal do tarso, ou seus ramos plantares, e se manifesta por dor, dormência e parestesia na planta do pé.
Síndrome de Turner . Síndrome caracterizada por agenesia gonadal e múltiplas anomalias de causa genética (monossomia do cromossomo X , tipo XO). Suas características são: a) baixa estatura e pescoço curto; b) linfedema de membros superiores e inferiores; c) agenesia gonadal com infantilismo sexual, genitália externa feminina, hipoplasia mamária e amenorréia primária (em mulheres), ed) diminuição dos estrogênios sanguíneos com aumento das gonadotrofinas na urina. O estudo da cromatina sexual revela que alguns desses pacientes são geneticamente "homens" e outros são geneticamente "mulheres", apesar de não haver diferenças fenotípicas entre eles.
Síndrome de Turner masculina . veja Síndrome de Noonan
Síndrome de Uehlinger . Síndrome caracterizada pela associação de hiperostose generalizada com espessamento localizado da pele. O quadro é definido pelo desenvolvimento de uma osteosc1erose difusa que afeta as epífises, metáfises e diáfises e que se manifesta por deposição subperiosteal de quantidades anormais de osso esponjoso devido à dor, inchaço e rigidez das articulações e por espessamento acentuado do pele do antebraço e antebraço. Começa na puberdade ou adolescência e a causa é desconhecida.
Síndrome unha-patela . Síndrome hereditária caracterizada por anormalidades nos ossos e nas unhas e doença renal. Suas características são: a) cotovelos em cunha, rótulas rudimentares e configuração de chifre dos ossos ilíacos; b) unhas deformadas com múltiplas fissuras ec) sinais de glomerulonefrite crônica moderada, geralmente de evolução benigna. O quadro é de uma osteosdisplasia, de causa desconhecida, e é transmitida como traço autossômico dominante.
Síndrome de Urbach-Wiethe . Síndrome de proteinose lipóide. O quadro é devido ao depósito extracelular de material hialino na pele e na mucosa da faringe e laringe, e se manifesta por: a) disfonia ou afonia, macroglossia e anomalias dentárias; b) desenvolvimento progressivo de pápulas cutâneas de distribuição generalizada, de coloração branco-amarelada e compostas por material intensamente positivo na reação PAS; ec) presença de calcificações simétricas na região da sela túrcica. É uma doença granulomatosa de armazenamento de lipídios que geralmente não impede uma vida longa e sua causa é desconhecida.
Síndrome urêmico-hemolítica . Síndrome de insuficiência renal aguda em lactentes associada a hemólise, distúrbios de coagulação e hemorragias. É uma condição grave da primeira infância e é caracterizada pela presença de hemólise intravascular, trombocitopenia, hemorragias cutâneas e mucosas e manifestações neurológicas, com fenômenos de coagulação intravascular e insuficiência renal aguda. A causa não foi estabelecida com precisão, mas é atribuída a uma reação atípica do corpo em resposta a diferentes infecções virais.
Síndrome de esvaziamento rápido . Síndrome de gastrectomia . Aparecimento de episódios pós-prandiais de náusea, transpiração, palpitações, fraqueza acentuada, sensação de calor em ondas e, às vezes, cólicas diarreicas, em indivíduos submetidos a gastrectomia parcial com gastrojejunostomia. É atribuída a um esvaziamento excessivamente rápido do conteúdo gástrico no jejuno.
Síndrome do infarto do miocárdio vagal . Síndrome dos enfartes da face inferior do coração. O quadro inclui distúrbios digestivos funcionais (soluços, arrotos, náuseas ou vômitos), fenômenos vasomotores (palidez, sudorese, hipotensão, lipotimia) e distúrbios do ritmo cardíaco (bradicardia sinusal ou nodal, bloqueio, sinoatrial, dissociação atrioventricular com interferências); em pacientes com infarto do miocárdio da face inferior. É visto com relativa frequência e responde rapidamente à administração de atropina.
Síndrome de Van Buchem . Síndrome de hiperostose cortical generalizada. Condição hereditária caracterizada por osteosclerose do crânio, mandíbula, clavículas, costelas e diáfise de ossos longos, com aumento da fosfatase alcalina sérica. Devido à progressiva compressão óssea, costuma ocorrer atrofia óptica, paralisia facial e surdez perceptual. É transmitido como um traço autossômico recessivo.
Síndrome da veia cava inferior . Síndrome de obstrução da veia cava inferior. Edema progressivo das extremidades inferiores e escroto, dilatação das veias superficiais das pernas e do abdome inferior, oligúria e às vezes ascite. É observada em condições graves, como peritonite generalizada e trombose ascendente das veias dos membros inferiores, e geralmente é uma complicação terminal.
Síndrome da veia cava superior . Síndrome de obstrução do fluxo da veia cava superior. É caracterizada por edema da face, pescoço, tórax superior e membros superiores (edema edema), ingurgitamento fixo das veias do pescoço, cianose predominantemente facial e hepatomegalia dolorosa. É devido à compressão extrínseca das paredes dos vasos, geralmente devido a um tumor.
Síndrome de Verner-Morrison . veja Síndrome de WDHA .
Síndrome de Vernet . Síndrome neurológica da base do crânio. É caracterizada por paralisia unilateral progressiva e simultânea, embora de grau variável, dos nervos cranianos IX, X e XI, devida à compressão exercida por neurinomas, focos de sarcoidose ou tumores infiltrantes da base do crânio.
Síndrome do vértice de Crag . veja síndrome de Gradenigo .
Síndrome de vertigem cervical . Síndrome neurológica devido à insuficiência da circulação vertebral. Episódios súbitos e transitórios de tontura, vertigem, nistagmo, ataxia e diplopia, ou combinações de alguns desses sintomas, que geralmente são desencadeados por movimentos forçados ou excessivamente amplos da cabeça e do pescoço. É causada por compressão ou espasmo da artéria vertebral
Síndrome de Villaret . Síndrome de lesão dos nervos cranianos IX, X, XI e XII, e do simpático cervical, por compressão tumoral. É unilateral e se caracteriza por: a) perda do paladar no terço posterior da língua (lesão do IX nervo craniano); b) paralisia do constritor superior da faringe, com dificuldade em engolir sólidos (nervo X); c) paralisia do palato mole e dos maxilares, com anestesia associada (nervo X); d) paralisia das cordas vocais (nervo X); e) paralisia do trapézio e esternocleidomastóideo (nervo XI); f) paralisia do meio da língua (nervo XII), eg) síndrome de Horner (paralisia simpática cervical).É produzida por tumores do corpo parotídeo e carotídeo, tumor ou adenopatias tuberculosas.
Síndrome de virilização adrenal em adultos . Variedade de síndrome adrenogenital. Suas manifestações, nas mulheres, são as de produção excessiva de andrógenos: hirsutismo, acne, seborréia, calvície temporária, aumento da massa e força muscular, amenorréia com atrofia uterina e hipertrofia do clitóris, masculinização da voz, etc. No homem adulto, obviamente, a imagem é mais difícil de detectar. A causa é frequentemente tumoral (adenomas adrenais ou carcinomas) e há um aumento acentuado na excreção urinária de 17-cetosteroides.
Síndrome de Vogt . Núcleo caudado e síndrome de lesão por putâmen. É caracterizada pela presença de movimentos atetósicos, oscilação rítmica dos membros e choro e risos espasmódicos e explosivos. Não há paralisia ou fenômenos sensíveis.
Síndrome de Vogt-Koyanagi . Síndrome caracterizada por alterações na pele e nos órgãos dos sentidos. É uma associação de vitiligo e áreas de alopecia, com distúrbios oculares e surdez, às vezes acompanhada de labirintite.
Síndrome de Volkmann . Síndrome de necrose e retração muscular devido a isquemia regional aguda. É uma condição isquêmica, decorrente da interrupção do fluxo arterial diretamente relacionada à compressão exercida pelos focos de fratura, ou pelos modelos de gesso para imobilizá-los, que geralmente atinge a mão e a superfície flexora do antebraço e se manifesta por dor intensa. , edema, cianose, parestesias e incapacidade de movimentar os dedos e, se não houver resolução rápida, por necrose muscular e retração cicatricial em flexão.
Síndrome de Waardenburg . Síndrome hereditária de indivíduos com surdez e anormalidades predominantemente faciais. Observam-se albinismo dos cílios e tufo de pelos brancos na região frontal, união das sobrancelhas na linha média, heterocromia da íris, nariz achatado e surdez congênita. É transmitido como um traço autossômico dominante.
Síndrome de Wallenberg . Síndrome neurológica do tronco cerebral com distúrbios sensoriais e cerebelares e envolvimento dos nervos cranianos V, IX, X e XI. É caracterizada, no lado da lesão, por anestesia corneana e paralisia dos músculos mastigatórios (lesão do nervo V); Dificuldade em engolir e perda do paladar no terço posterior da língua (nervo IX); paralisia do palato mole e cordas vocais (nervo X); paralisia do trapézio e esternocleidomastóideo (nervo Xl); Síndrome de Horner (lesão da via pupilodilatadora descendente) e ataxia cerebelar (vias espinocerebelares e olivocerebelares);e no lado oposto devido à perda de sensibilidade térmica e dor (feixe espinotalâmico lateral). A imagem é produzida pela oclusão da artéria cerebelar posteroinferior.
Síndrome de Waterhouse-Friderichsen . Síndrome da forma fulminante de meningite cerebrospinal. É um quadro de início súbito e evolução rápida, caracterizado por palidez, dispneia, cianose, febre alta com calafrios, petéquias e equimoses na pele e mucosas, colapso e coma. É observada em pacientes pediátricos e é consequência de uma sepse muito grave, geralmente causada por meningococos ou pneumococos.
Síndrome de WDHA . Síndrome de tumores pancreáticos produtores de diarreia. O nome deriva das iniciais; Inglês de suas manifestações características: diarréia aquosa (diarréia aquosa), hipocalemia (hipocalemia) e aclorídrica (acloridria). A diarreia é geralmente abundante, grave o suficiente para às vezes levar ao choque ou produzir hipocalemia alarmante; há uma diminuição na secreção de ácido gástrico basal, e a hipercalcemia e a hiperglicemia estão frequentemente presentes. A condição é produzida por tumores não insulínicos das ilhotas de Langerhams e suas manifestações são aparentemente devidas à hipersecreção de polipeptídeos intestinais vasoativos.
Síndrome de Weber . Síndrome neurológica do tronco cerebral com hemiplegia alternada e envolvimento do terceiro nervo craniano. A imagem é de uma hemiplegia do lado oposto ao da lesão, com ptose palpebral, estrabismo e perda dos reflexos de luz e acomodação do lado da lesão. É típico de lesões, quase sempre vasculares, que acometem o feixe piramidal do pé do pedúnculo cerebral e as fibras radiculares do terceiro nervo craniano.
Síndrome de Weber-Christian . Síndrome da paniculite nodular não supurativa. É caracterizada pela formação de nódulos dolorosos recorrentes no tecido adiposo subcutâneo, com febre e alteração do estado geral; os nódulos evoluem deixando uma depressão central. O prognóstico é benigno e a causa desconhecida.
Síndrome de Wegener . Síndrome da granulomatose respiratória necrosante. É caracterizada por vasculite necrosante e infecção granulomatosa do trato respiratório e rins, e freqüentemente de outros órgãos. Há cefaleia, febre, artralgia, sinusite, otite, tosse, hemoptise, etc. Predominam na meia-idade e é considerada relacionada a fenômenos de hipersensibilidade.
Síndrome de Weill-Marchesani . Síndrome da distrofia mesodérmica congênita. É caracterizada por: a) baixa estatura com braquicefalia e braquidactilia; b) tórax largo com massas; músculos desenvolvidos que dão a impressão de grande força corporal; c) alterações oculares, com ectopia do cristalino e frequentemente glaucoma e miopia; d) rigidez articular acentuada e, ocasionalmente, e) estenose pulmonar congênita. É transmitido como traço autossômico recessivo.
Síndrome de Werdnig-Hoffmann . Síndrome da artrofia muscular espinhal familiar . É caracterizada pela instalação precoce (primeira infância) de hipotonia e atrofia muscular, seguida de paralisia flácida completa e finalmente morte. É devido a uma degeneração das células do corno anterior da medula e é transmitida como um traço autossômico recessivo.
Síndrome de Wermer . Síndrome de neoplasias endócrinas múltiplas tipo 1 . É definida pela presença de tumores ou hiperplasia da paratireóide, tireóide, glândulas adrenais, ilhotas de Langerhans e hipófise, e por um quadro clínico dependente dos sistemas; hiperfuncional no momento em que o indivíduo consulta. Podem ser observadas combinações das seguintes condições: a) hipoglicemia; b) hipercalcemia com nefrocalcinose; c) úlceras pépticas múltiplas (síndrome de Zollinger-Ellison); d) múltiplos lipomas da pele;e) distúrbios produzidos pela expansão de um tumor hipofisário ou por disfunção desta glândula (cefaleia, perda gradual de visão, amenorreia secundária, etc.) e, menos frequentemente, síndrome de Cushing, hepatomegalia e outras manifestações.
Síndrome de Werner . Síndrome hereditária com alterações cutâneas e tendência ao desenvolvimento de processos malignos. É caracterizada por envelhecimento prematuro, catarata, distúrbios semelhantes à esclerodermia, ulcerações nas pernas e aterosclerose, com tendência aumentada para o desenvolvimento de sarcomas e meningiomas.
Síndrome de Wernicke . Síndrome de presbiofrenia. É caracterizada por fraqueza mental com falta de fixação e desorientação, ansiedade, alucinações, delírios e outros transtornos. É uma psicopatia típica da velhice.
Síndrome de Weyers-Thier . Síndrome da displasia oculovertebral. É caracterizada por múltiplas anormalidades oculares (microftalmia, anoftalmia, etc.), assimetria facial, hipoplasia frontal e displasia orbitária e malformações da coluna vertebral e da inserção das costelas. É devido a um distúrbio no início do desenvolvimento do embrião e afeta apenas os homens.
Síndrome de Wilfred Harris . Síndrome de irritação do nervo craniano IX. Ataques intensos de dor paroxística localizada na garganta e, às vezes, irradiando para o ouvido. Repete-se com periodicidade variável, às vezes sem motivo aparente e outras; desencadeada pela deglutição. A etiologia é desconhecida, embora do ponto de vista clínico o quadro seja em todos os aspectos comparável ao da neuralgia do trigêmeo.
Síndrome de Wilson-Mikity . Síndrome clínica e radiológica de imaturidade pulmonar. É a associação de uma cianose persistente no recém-nascido, com manifestações radiológicas características: múltiplos focos de enfisema localizado, de 1 a 10 mm de diâmetro, circundados por uma faixa de condensação. Outras características são sua apresentação em prematuros, com muito baixo peso ao nascer, a ausência de sinais de infecção, seu curso lento, alta mortalidade e a possibilidade de recuperação completa nos que sobrevivem.
Síndrome de Wolff-Parkinson-White . Síndrome eletrocardiográfica e clínica de excitação atrioventricular anormal. É definida pela presença, no eletrocardiograma de uma onda P normal, um PR curto (menos de 0,11 seg) e um QRS alargado com preenchimento de sua porção inicial (onda delta) anormalidade que está associada a uma tendência franca ao desenvolvimento de episódios de taquicardia atrial paroxística ou fibrilação atrial. É observada em jovens sem outros sinais de cardiopatia e se deve à existência de vias de condução acessórias paralelas ao feixe de His.
Síndrome de Woltman-Moersch . S tetaniforme índrome de causa não infecciosa. É observada na idade adulta, sem distinção entre os sexos, e é caracterizada por: a) o aparecimento de contrações espasmódicas intermitentes, de distribuição irregular, que predominam nos músculos do tronco e extremidades; b) estacionamentos prolongados nestas condições, ou progressão lenta com aumento dos grupamentos musculares afetados e maior duração das contrações; ec) neste último caso, evolução para um estado de virtual rigidez generalizada. A causa é desconhecida, embora seja atribuída a uma deficiência adquirida de mediadores fisiológicos que inibem a atividade dos neurônios motores.
Síndrome de Wunderlich . Síndrome de hematoma perirrenal espontâneo. Manifesta-se abruptamente por lombalgia unilateral intensa, sinais de anemia aguda e aparecimento rápido de tumor lombar palpável, além de náuseas e vômitos, meteorismo por íleo reflexo, contratura lombar e oligúria. Pode aparecer no curso renal (pielonefrite, tumores, etc.), hemático (hemofilia, leucemia) ou vascular (periarterite, trombose de cava, etc.).
Síndrome de Zollinger-Ellison . Síndrome do tumor pancreático ulcerogênico. É manifestada por uma tríade característica: úlceras pépticas intratáveis, às vezes fulminantes, hiperacidez gástrica extrema e diarreia aquosa ou esteatorreia. É causada pela presença de tumores não beta das ilhotas de Langerhans, únicos ou múltiplos, benignos ou malignos, e atualmente é considerada tipo I de neoplasias endócrinas múltiplas.